
Acerca del autor

Mohamed Jobran
Consultor Regulatorio (licenciatura en Farmacia, maestría en Informática Biomédica). Con 19 años de experiencia trabajando para SFDA, empresas multinacionales y como consultor profesional.
Navegar por el proceso de registro de dispositivos médicos SFDA en Arabia Saudita, particularmente el proceso de Autorización de Comercialización de Dispositivos Médicos (MDMA), es esencial para los fabricantes que buscan ingresar al mercado saudí en rápida expansión.
Los requisitos de la MDMA han evolucionado para adaptarse al avance de las normas internacionales, como la evolución de las Directivas de Productos Sanitarios de la UE (MDD) a los Reglamentos de Productos Sanitarios (MDR). El continuo desarrollo de la normativa sobre productos sanitarios en Arabia Saudí ha incrementado la complejidad de este proceso.
Por ejemplo, incluso los dispositivos médicos de bajo riesgo (suministros y consumibles) comenzaron a necesitar un representante autorizado (AR) y la aprobación de MDMA.
En PharmaKnowl, ofrecemos asistencia experta para el proceso de registro. Tanto si se trata de nuevos dispositivos como de la modificación de productos existentes, nuestro equipo proporciona la visión estratégica y la experiencia técnica necesarias para lograr una entrada oportuna y exitosa al mercado de Arabia Saudita.
Este artículo aclara los requisitos de registro de dispositivos médicos SFDA y define sus diferencias de clasificación.
Para otros tipos de registro de productos, consulte la publicación de registro de SFDA .
Tabla de contenido
Solicitudes de registro
La SFDA regula los productos sanitarios en Arabia Saudita para garantizar su seguridad, eficacia y calidad. Los fabricantes deben obtener la aprobación de MDMA antes de comercializar sus productos. Tras la aprobación, son responsables de los informes de seguridad y otras operaciones de gestión del ciclo de vida, como la actualización, la renovación y la presentación de la Identificación Única del Dispositivo (UDI) .
A continuación se presenta un resumen de los procedimientos de registro de dispositivos médicos:
MDNR (riesgo bajo) – Cancelado
Las empresas solían registrar dispositivos médicos no estériles, sin medición y de bajo riesgo a través del Registro Nacional de Dispositivos Médicos (MDNR), también conocido como «Lista de Dispositivos Médicos», que está exento de MDMA y AR. Sin embargo, en septiembre de 2022, la SFDA canceló el procedimiento MDNR . Por lo tanto, todos los dispositivos deben contar con un expediente técnico para MDMA con un AR designado en Arabia Saudita.
MDMA 1 (GHTF) – Cancelada
En esta vía de solicitud, la SFDA consideró las aprobaciones de los países miembros del GHTF, como la Unión Europea, Estados Unidos, Canadá, Australia y Japón. Era una vía fácil de seguir, sin necesidad de realizar consultas técnicas exhaustivas. Sin embargo, a finales de 2021, la SFDA había cancelado esta vía del GHTF.
MDMA 2 (TFA)
Desde enero de 2022, todos los productos sanitarios y diagnósticos in vitro (IVD) deben obtener una autorización SFDA a través de la vía de registro de la Solicitud de Expediente Técnico (TFA) (MDMA2). Esta vía es el resultado de las continuas actualizaciones regulatorias de la autoridad, influenciadas por el MDR y el IVDR de la UE. Requiere presentar la documentación técnica de forma clara, consultable y organizada.
Por lo tanto, los requisitos de registro se volvieron más estrictos. Se deben cumplir numerosas condiciones y presentar estudios, como informes de evaluación clínica (CER), informes de pruebas de biocompatibilidad y la necesidad de realizar un estudio de seguimiento clínico poscomercialización (PMCF) en Arabia Saudita.
Aunque los requisitos del TFA son similares a los de la UE (marcado CE), la SFDA no exige un certificado CE. Sin embargo, si el expediente del producto no lleva el marcado CE, se someterá a una evaluación rigurosa.
Requisitos de registro
A continuación se resumen los requisitos, que varían según el tipo y la clase de producto. A continuación, se presentan las secciones principales del expediente de registro:
- Tabla de contenidos
- Descripción del dispositivo
- Uso previsto/finalidad
- Historial del dispositivo
- Clasificación de dispositivos.
- Etiqueta del dispositivo
- Modelos de dispositivos, accesorios y variantes
- Instrucciones de uso
- Información de diseño
- Información de fabricación
- Principios esenciales (lista de principios esenciales obligatorios) de seguridad y rendimiento
- Análisis beneficio-riesgo
- Expediente de Gestión de Riesgos (Plan e Informe)
- Pruebas preclínicas
- Informes de pruebas
- Informes de pruebas de biocompatibilidad.
- Plan e informe de investigación clínica
- Informe de evaluación clínica (CER)
- Seguimiento clínico posterior a la comercialización (PMCF)
- Vigilancia posterior a la comercialización (PMS), plan y reporte
- Informe periódico de actualización de seguridad (PSUR) para dispositivos médicos.
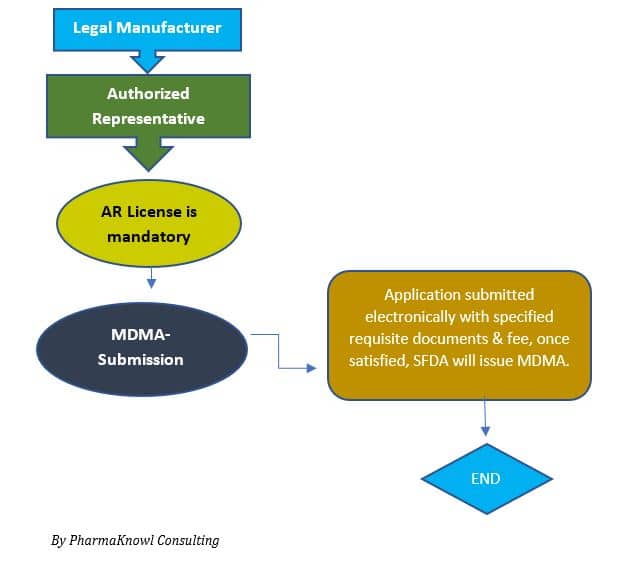
Proceso de Registro
Time needed: 90 days
Podemos resumir el proceso de aprobación del registro de dispositivos médicos (MDMA) en los siguientes pasos:
- Designación de un representante autorizado (AR)
Los fabricantes deben designar un AR para representar a su empresa en Arabia Saudita.
- Análisis de brechas
Las empresas deben realizar un análisis exhaustivo de las deficiencias del expediente de registro frente a todos los requisitos aplicables. Identificar las deficiencias es esencial para determinar los estudios y pruebas pertinentes, como estabilidad, CER, biocompatibilidad, gestión de riesgos y seguimiento clínico poscomercialización (PMCF).
- Presentación de MDMA
El solicitante presenta el expediente técnico a la SFDA .
- Validación
SFDA garantiza buenas prácticas de presentación sin evaluar su contenido.
- Pago de tasas
El solicitante recibirá una factura SFDA antes de la evaluación.
- Evaluación de la aplicación
Los evaluadores revisarán el expediente y enviarán sus consultas en varias oleadas.
- Aprobación de MDMA
Los fabricantes legales reciben un certificado MDMA que les permite comercializar el producto en Arabia Saudita.
Clasificación
Los solicitantes deben clasificar sus productos según las normas de clasificación de dispositivos médicos SFDA en la solicitud de MDMA. Estas normas se corresponden con el reglamento de la UE (certificado CE), ya que constituyen el reglamento de referencia no oficial SFDA , pero no necesariamente coinciden con él.
Dispositivo médico
La clasificación de dispositivos médicos SFDA es Clase A, B, C o D. Se basa en su clase de riesgo. La clase es necesaria para determinar el procedimiento de registro y sus requisitos, como se describe en la siguiente tabla:
| Clasificación SFDA | Clase de riesgo | Regla de clasificación MDR |
| Clase A | Bajo | I |
| Clase A – Estéril | Bajo-medio | Es |
| Clase A – Función de medición | Bajo-medio | Soy |
| Clase A – Instrumentos quirúrgicos reutilizables | Bajo-medio | Ir |
| Clase B | Bajo-medio | IIa |
| Clase C | Medio-alto | IIb |
| Clase D | Alto | III |
La clasificación es según lo siguiente:
- Uso previsto.
- Nivel de riesgo (probabilidad y gravedad del daño a los pacientes, usuarios y otros).
- Invasividad del cuerpo humano.
- Duración de uso.
Dispositivos idénticos podrían clasificarse de forma diferente según la parte del cuerpo a la que se dirigen. Por lo tanto, el uso previsto es crucial para determinar la clasificación correcta. La referencia al uso previsto se refleja en lo siguiente:
- Instrucciones de uso (IFU)
- Etiqueta
- Materiales promocionales
- Expediente técnico
Diagnóstico in vitro (IVD)
En lo que respecta al diagnóstico in vitro, la SFDA también está adoptando el reglamento europeo de dispositivos médicos IVDR:
| Clasificación IVD SFDA | Clase de riesgo | Regla de clasificación |
| Clase A | Bajo riesgo individual y bajo riesgo para la salud pública | A |
| Clase B | Riesgo individual moderado y/o riesgo bajo para la salud pública | B |
| Clase C | Alto riesgo individual y/o riesgo moderado para la salud pública | do |
| Clase D | Alto riesgo individual y alto riesgo para la salud pública | D |
El solicitante es responsable de determinar la clase del IVD mediante:
- Aplicación de las reglas de clasificación para los dispositivos médicos de diagnóstico in vitro, y
- En vista de:
- Uso previsto
- Nivel de riesgo
Agrupación de varios dispositivos
Se permite agrupar varios dispositivos en la misma solicitud; el solicitante puede agrupar hasta 50 artículos en una sola solicitud de MDMA. Sin embargo, los productos deben cumplir varias condiciones, como tener la misma clase de riesgo y el mismo uso previsto.
Líneas de tiempo
Los plazos de evaluación varían según diversos factores, como la clase de riesgo, la complejidad del producto, el número de productos agrupados y la integridad del expediente. La falta de identificación de deficiencias antes de la presentación podría retrasar la aprobación. Los plazos completos del proyecto de registro incluyen:
- Tiempo de análisis de brechas
Esta parte depende de la cantidad y el tipo de espacios, la velocidad de la respuesta del fabricante y la experiencia del personal regulador. - Tiempo de evaluación SFDA
Para conocer los plazos de revisión oficiales, consulte nuestro artículo Plazos SFDA .
Honorarios
Las tarifas de solicitud de MDMA varían según la clase de riesgo del dispositivo médico y la cantidad de productos incluidos en la solicitud. Para más información, consulte nuestra publicación actualizada sobre tarifas SFDA .
Renovación
La validez predeterminada de la MDMA es de tres años. Cuando la licencia está próxima a vencer, los fabricantes pueden presentar una solicitud de renovación de MDMA hasta tres meses (90 días) antes de la fecha de vencimiento. Si se registra en la obsoleta solicitud MDMA1 (GHTF), el proceso de renovación sería largo y requeriría requisitos extensos, al igual que un nuevo registro. En cambio, la renovación de la MDMA2 será más rápida.
Actualizar
Para realizar cambios en el dispositivo médico registrado, los fabricantes deben presentar una solicitud de actualización de MDMA .
Certificado MDMA
SFDA emitirá un certificado MDMA en árabe e inglés que contendrá lo siguiente:
- Información del fabricante (titular de la licencia)
- Nombre del dispositivo médico e información del grupo de dispositivos médicos.
- Periodo de validez
- Número de certificado

Categorías de productos
A continuación se muestran ejemplos de tipos y categorías de dispositivos médicos en Arabia Saudita:
- Dispositivos de diagnóstico:
- Equipos de imagen: máquinas de rayos X, escáneres CT.
- Dispositivos de monitorización: aparatos de ECG y monitores de presión arterial.
- Dispositivos de diagnóstico in vitro: pruebas de embarazo, medidores de glucosa en sangre.
- Dispositivos terapéuticos:
- Dispositivos respiratorios: ventiladores, nebulizadores.
- Dispositivos de infusión: bombas de insulina y equipos de terapia intravenosa.
- Dispositivos de rehabilitación: Sillas de ruedas, prótesis.
- Dispositivos implantables:
- Dispositivos cardiovasculares: marcapasos, stents.
- Implantes ortopédicos: reemplazos de cadera, barras espinales, reemplazos de articulaciones.
- Instrumentos quirúrgicos:
- Instrumentos de corte: Bisturís, tijeras quirúrgicas, fórceps.
- Grapadoras y Suturas: Grapadoras quirúrgicas, suturas absorbibles.
- Dispositivos dentales:
- Materiales restauradores: Coronas dentales, empastes.
- Aparatos de Ortodoncia: Brackets, retenedores.
- Otros: taladros dentales
- Dispositivos oftálmicos:
- Lentes Correctivos: Lentes de contacto, gafas.
- Equipo Quirúrgico: Máquinas LASIK, lentes intraoculares.
- Software médico (SaMD) :
- Software de diagnóstico: IA para análisis de imágenes.
- Software de planificación de tratamiento: herramientas de planificación de radioterapia.
- Equipo de laboratorio:
- Analizadores de gases en sangre y analizadores de hematología.
- Equipos de esterilización: Autoclaves, esterilizadores UV.
- Gases medicinales
- Dispositivos para el cuidado de la salud en el hogar:
- Dispositivos de monitoreo: Monitores de glucosa en sangre y termómetros digitales.
- Ayudas a la movilidad: Andadores, patinetes eléctricos.
- Dispositivos terapéuticos (por ejemplo, máquinas de diálisis)
Licencia de importación de dispositivos médicos (MDIL)
La importación de dispositivos médicos requiere MDMA válido. Sin embargo, la SFDA puede eximir del registro a algunos dispositivos médicos y solo exigirles la emisión de una licencia de importación de dispositivos médicos (MDIL). Por ejemplo:
- Dispositivos médicos con fines de demostración o formación.
- Productos químicos (producto terminado), ya sean clasificados como dispositivo médico o utilizados junto con dispositivos médicos (por ejemplo, gases utilizados para calibrar dispositivos médicos, así como productos químicos utilizados para garantizar la esterilización de dispositivos médicos, la fabricación de prótesis y la conservación de tejidos o células). Se excluyen los siguientes productos (si el producto químico está clasificado como dispositivo médico y el dispositivo debe contener MDMA).
- Materiales radioactivos
- Diagnóstico in vitro médico
- Diagnóstico in vitro no médico
- Precursores químicos.
- Los aparatos de destilación para proveedores de atención médica o instalaciones educativas.
- Productos para uso investigativo y educativo.
- Dispositivos y suministros médicos semiacabados (y productos químicos crudos y no crudos) para fabricación local (la fabricación incluye reacondicionamiento, ensamblaje, empaquetado y etiquetado).
Registro de empresa
La SFDA también regula a las empresas de dispositivos médicos, aplicando diferentes requisitos para empresas locales e internacionales. A continuación, se detallan los requisitos para ambos tipos:
Compañía local saudí
La SFDA otorga licencias a empresas locales de dispositivos médicos, como importadores, distribuidores, almacenes, representantes autorizados y fabricantes. La licencia local se denomina Licencia de Establecimiento de Dispositivos Médicos (MDEL). Todas las empresas mencionadas anteriormente deben implementar un sistema de gestión de calidad (SGC) y contar con la certificación ISO 13485 .
Fabricante legal
Los fabricantes internacionales con licencia deben designar un representante autorizado (RA) en Arabia Saudita; este es el primer paso para facilitar la comunicación con la SFDA . La principal responsabilidad del RA es la conformidad del producto en las actividades previas y posteriores a la comercialización. El RA mantendrá el estado del registro, facilitará los envíos, supervisará la seguridad, informará sobre los casos y representará a la empresa en asuntos regulatorios o legales.
Servicio de Registro
Ya sea que su empresa sea nueva o ya esté activa en el mercado saudí, PharmaKnowl lo ayudará a cumplir con la SFDA y facilitará su negocio al brindar cumplimiento, información y gestión de suministros a sus distribuidores sin comisión.
PharmaKnowl es un proveedor de servicios con licencia SFDA en Arabia Saudita que representa a reconocidas empresas internacionales. Contáctenos para programar una reunión exploratoria o solicitar más información.



