
Über den Autor

Mohammed Jobran
Regulierungsberater (Apotheker BSc, Biomedizinische Informatik MSc). Mit 19 Jahren Erfahrung in der Arbeit für die SFDA, multinationale Unternehmen und als professioneller Berater.
Für Hersteller, die in den schnell wachsenden saudischen Markt einsteigen möchten, ist es von entscheidender Bedeutung, den Registrierungsprozess für Medizinprodukte SFDA in Saudi-Arabien zu meistern, insbesondere den Prozess der Marktzulassung von Medizinprodukten (Medical Device Marketing Authorization, MDMA).
Die MDMA-Anforderungen haben sich an die Weiterentwicklung internationaler Standards angepasst, beispielsweise an die Entwicklung der EU-Richtlinien für Medizinprodukte (MDD) zur Medizinprodukteverordnung (MDR). Die kontinuierliche Weiterentwicklung der Medizinproduktevorschriften in Saudi-Arabien hat die Komplexität dieses Prozesses erhöht.
Beispielsweise waren selbst für medizinische Geräte (Zubehör und Verbrauchsmaterialien) mit geringem Risiko inzwischen eine Zulassung durch einen autorisierten Vertreter (AR) und eine MDMA-Zulassung erforderlich.
Wir von PharmaKnowl bieten Ihnen kompetente Unterstützung im Registrierungsprozess. Ob Sie neue Geräte entwickeln oder bestehende Produkte modifizieren – unser Team bietet Ihnen die strategischen Einblicke und das technische Know-how für einen zeitnahen und erfolgreichen Markteintritt in Saudi-Arabien.
Dieser Artikel erläutert die Registrierungsanforderungen für Medizinprodukte SFDA und definiert ihre Klassifizierungsunterschiede.
Informationen zu anderen Arten der Produktregistrierung finden Sie im Registrierungsbeitrag SFDA .
Inhaltsverzeichnis
Registrierungsanträge
Die SFDA reguliert Medizinprodukte in Saudi-Arabien, um deren Sicherheit, Wirksamkeit und Qualität zu gewährleisten. Die Hersteller müssen vor der Vermarktung ihrer Produkte eine MDMA-Zulassung einholen. Nach der Zulassung sind sie für die Sicherheitsberichterstattung und andere Lebenszyklusmanagement-Maßnahmen wie Aktualisierung, Erneuerung und Einreichung der Unique Device Identification (UDI) verantwortlich.
Hier ist eine Zusammenfassung der Registrierungsverfahren für Medizinprodukte:
MDNR (geringes Risiko) – Abgesagt
Unternehmen registrierten unsterile, nicht messende Medizinprodukte mit geringem Risiko bisher über das Medical Device National Registry (MDNR), auch bekannt als „Medical Devices Listing“, das von MDMA und AR ausgenommen ist. Im September 2022 hat die SFDA das MDNR-Verfahren jedoch eingestellt . Daher müssen alle Produkte über eine technische Akte für MDMA bei einem zuständigen AR in Saudi-Arabien verfügen.
MDMA 1 (GHTF) – Abgesagt
Bei diesem Antragsverfahren berücksichtigte die SFDA Zulassungen aus den GHTF-Mitgliedsländern wie der Europäischen Union, den Vereinigten Staaten, Kanada, Australien und Japan. Es war ein einfacher Weg, der ohne tiefgreifende technische Fragen möglich war. Ende 2021 hatte die SFDA dieses GHTF-Verfahren jedoch eingestellt.
MDMA 2 (TFA)
Seit Januar 2022 müssen alle Medizinprodukte und In-vitro-Diagnostika (IVDs) eine Zulassung SFDA über das Registrierungsverfahren Technical File Application (TFA) (MDMA2) erhalten. Dieses Verfahren entstand aufgrund der kontinuierlichen Regulierungsaktualisierungen der Behörde unter dem Einfluss der EU-MDR und IVDR. Es erfordert eine klare, durchsuchbare und strukturierte Darstellung der technischen Dokumentation.
Daher wurden die Registrierungsanforderungen verschärft. Zahlreiche Bedingungen müssen erfüllt und Studien vorgelegt werden, wie z. B. klinische Bewertungsberichte (CER), Berichte über Biokompatibilitätstests und die Notwendigkeit, eine klinische Nachbeobachtungsstudie (PMCF) nach der Markteinführung in Saudi-Arabien durchzuführen.
Obwohl die TFA-Anforderungen denen der EU (CE-Kennzeichnung) ähneln, verlangt die SFDA kein CE-Zertifikat. Trägt die Produktakte jedoch keine CE-Kennzeichnung, wird sie einer strengen Prüfung unterzogen.
Registrierungsvoraussetzungen
Im Folgenden sind die Anforderungen zusammengefasst, die je nach Produkttyp und -klasse variieren. Hier sind die wichtigsten Abschnitte der Registrierungsdatei:
- Inhaltsverzeichnis
- Gerätebeschreibung
- Verwendungszweck
- Geräteverlauf
- Geräteklassifizierung.
- Geräteetikett
- Gerätemodelle, Zubehör und Varianten
- Gebrauchsanweisung
- Konstruktionsinformationen
- Herstellungsinformationen
- Grundlegende Prinzipien (EP-Liste) für Sicherheit und Leistung (Checkliste der erforderlichen Grundvoraussetzungen)
- Nutzen-Risiko-Analyse
- Risikomanagementdatei (Plan und Bericht)
- Präklinische Tests
- Prüfberichte
- Berichte zum Biokompatibilitätstest.
- Klinischer Prüfplan und Bericht
- Klinischer Bewertungsbericht (CER)
- Klinische Nachbeobachtung nach der Markteinführung (PMCF)
- Post Market Surveillance (PMS), Planen und Berichten
- Periodischer Sicherheitsupdatebericht (PSUR) für Medizinprodukte.
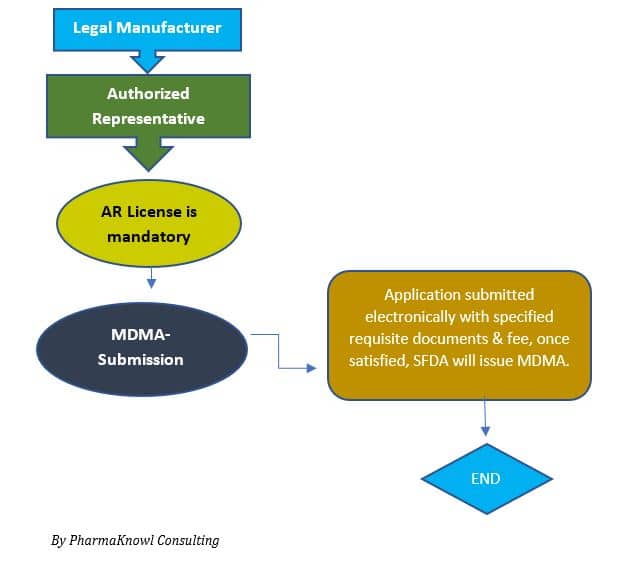
Registrierungsprozess
Benötigte Zeit: 90 Tage
Wir können den Genehmigungsprozess für die Registrierung von Medizinprodukten (MDMA) in den folgenden Schritten zusammenfassen:
- Ernennung eines Bevollmächtigten
Hersteller müssen einen AR ernennen, der ihr Unternehmen in Saudi-Arabien vertritt.
- Lückenanalyse
Unternehmen müssen eine gründliche Lückenanalyse der Registrierungsunterlagen im Hinblick auf alle geltenden Anforderungen durchführen. Die Identifizierung der Lücken ist entscheidend für die Bestimmung der relevanten Studien und Tests wie Stabilität, CER, Biokompatibilität, Risikomanagement und PMCF.
- MDMA-Einreichung
Der Antragsteller reicht die technischen Unterlagen bei der SFDA ein.
- Validierung
SFDA gewährleistet eine gute Einreichungspraxis, ohne den Inhalt zu bewerten.
- Gebührenzahlung
Der Antragsteller erhält vor der Beurteilung eine Rechnung SFDA .
- Bewerbungsbeurteilung
Die Gutachter prüfen die Akte und senden ihre Anfragen in mehreren Wellen zurück.
- MDMA-Zulassung
Die legalen Hersteller erhalten ein MDMA-Zertifikat, das ihnen die Vermarktung des Produkts in Saudi-Arabien erlaubt.
Einstufung
Antragsteller müssen ihre Produkte im Rahmen des MDMA-Antrags gemäß den Klassifizierungsregeln der SFDA für Medizinprodukte klassifizieren. Diese Regeln entsprechen der EU-Verordnung (CE-Zertifikat), da sie die inoffizielle Referenzverordnung SFDA darstellen, stimmen aber nicht unbedingt mit dieser überein.
Medizinprodukt
Die Klassifizierung von Medizinprodukten SFDA erfolgt in den Klassen A, B, C oder D. Sie richtet sich nach der jeweiligen Risikoklasse. Die Klasse ist für das Registrierungsverfahren und dessen Anforderungen entscheidend, wie in der folgenden Tabelle erläutert:
| SFDA -Klassifizierung | Risikoklasse | MDR Klassifizierungsregel |
| Klasse A | Niedrig | ICH |
| Klasse A – Steril | Niedrig-mittel | Ist |
| Klasse A – Messfunktion | Niedrig-mittel | Ich bin |
| Klasse A – Wiederverwendbare chirurgische Instrumente | Niedrig-mittel | Ir |
| Klasse B | Niedrig-mittel | IIa |
| Klasse C | Mittelhoch | IIb |
| Klasse D | Hoch | Drittes Kapitel |
Die Einteilung erfolgt nach den folgenden Kriterien:
- Verwendungszweck.
- Risikostufe (Wahrscheinlichkeit und Schwere des Schadens für Patienten, Benutzer und andere).
- Invasivität des menschlichen Körpers.
- Nutzungsdauer.
Identische Produkte können je nach Zielkörperteil unterschiedlich klassifiziert sein. Daher ist der Verwendungszweck entscheidend für die korrekte Klassifizierung. Der Verwendungszweck wird im Folgenden wiedergegeben:
- Gebrauchsanweisung (IFU)
- Etikett
- Werbematerialien
- Technisches Dokument
In-vitro-Diagnostik (IVDs)
Im Bereich der In-vitro-Diagnostik übernimmt die SFDA zudem die europäische Medizinprodukteverordnung IVDR:
| SFDA IVD-Klassifizierung | Risikoklasse | Klassifizierungsregel |
| Klasse A | Geringes individuelles Risiko und geringes Risiko für die öffentliche Gesundheit | A |
| Klasse B | Mäßiges individuelles Risiko und/oder geringes Risiko für die öffentliche Gesundheit | B |
| Klasse C | Hohes individuelles Risiko und/oder mäßiges Risiko für die öffentliche Gesundheit | C |
| Klasse D | Hohes individuelles Risiko und hohes Risiko für die öffentliche Gesundheit | D |
Die Bestimmung der IVD-Klasse obliegt dem Antragsteller durch:
- Anwendung der Klassifizierungsregeln für IVD-Medizinprodukte und
- Angesichts:
- Anwendungsgebiete
- Risikograd
Gruppieren mehrerer Geräte
Die Gruppierung mehrerer Geräte in einem Antrag ist zulässig; der Antragsteller kann bis zu 50 Artikel in einem MDMA-Antrag bündeln. Die Produkte müssen jedoch bestimmte Bedingungen erfüllen, beispielsweise die gleiche Risikoklasse und den gleichen Verwendungszweck aufweisen.
Zeitleisten
Die Bewertungszeiträume variieren je nach verschiedenen Faktoren, wie z. B. Risikoklasse, Komplexität des Produkts, Anzahl der gebündelten Produkte und Vollständigkeit der Akte. Nicht identifizierte Lücken vor der Einreichung können die Genehmigung verzögern. Die gesamten Zeitpläne für das Registrierungsprojekt umfassen:
- Lückenanalysezeit
Dieser Teil hängt von der Anzahl und Art der Lücken, der Geschwindigkeit des Hersteller-Feedbacks und der Erfahrung des Regulierungspersonals ab. - SFDA -Bewertungszeit
Die offiziellen Prüfungszeitpläne finden Sie in unserem Artikel „Zeitpläne SFDA .
Gebühren
Die MDMA-Antragsgebühren variieren je nach Risikoklasse des Medizinprodukts und der Anzahl der im Antrag enthaltenen Produkte. Weitere Informationen finden Sie in unserem aktualisierten Beitrag SFDA -Gebühren .
Erneuerung
Die Standardgültigkeit von MDMA beträgt drei Jahre. Wenn die Lizenz bald abläuft, können Hersteller bis zu drei Monate (90 Tage) vor Ablauf einen Antrag auf MDMA-Verlängerung stellen. Bei einer Registrierung im veralteten MDMA1-Antrag (GHTF) wäre der Verlängerungsprozess langwierig und würde, genau wie eine Neuregistrierung, umfangreiche Anforderungen erfordern. Im Gegensatz dazu ist die MDMA2-Verlängerung schneller.
Aktualisieren
Um Änderungen am registrierten Medizinprodukt vorzunehmen, müssen Hersteller einen MDMA-Update -Antrag einreichen.
MDMA-Zertifikat
SFDA stellt ein MDMA-Zertifikat in arabischer und englischer Sprache aus, das Folgendes enthält:
- Die Angaben des Herstellers (Lizenzinhaber)
- Name des Medizinprodukts und Informationen zur Medizinproduktgruppe.
- Gültigkeitsdauer
- Zertifikatsnummer

Produktkategorien
Hier sind Beispiele für Arten und Kategorien medizinischer Geräte in Saudi-Arabien:
- Diagnosegeräte:
- Bildgebende Geräte: Röntgengeräte, CT-Scanner.
- Überwachungsgeräte: EKG-Geräte und Blutdruckmessgeräte.
- In-vitro-Diagnostikgeräte: Schwangerschaftstests, Blutzuckermessgeräte.
- Therapeutische Geräte:
- Beatmungsgeräte: Beatmungsgeräte, Vernebler.
- Infusionsgeräte: Insulinpumpen und IV-Therapiegeräte.
- Rehabilitationsgeräte: Rollstühle, Prothesen.
- Implantierbare Geräte:
- Herz-Kreislauf-Geräte: Herzschrittmacher, Stents.
- Orthopädische Implantate: Hüftersatz, Wirbelsäulenstäbe, Gelenkersatz.
- Chirurgische Instrumente:
- Schneidinstrumente: Skalpelle, chirurgische Scheren, Pinzetten.
- Klammergeräte und Nähte: Chirurgische Klammergeräte, resorbierbare Nähte.
- Dentalgeräte:
- Restaurationsmaterialien: Zahnkronen, Füllungen.
- Kieferorthopädische Geräte: Zahnspangen, Retainer.
- Sonstiges: Zahnbohrer
- Ophthalmische Geräte:
- Korrekturlinsen: Kontaktlinsen, Brillen.
- Chirurgische Ausrüstung: LASIK-Geräte, Intraokularlinsen.
- Medizinische Software (SaMD) :
- Diagnosesoftware: KI zur Bildanalyse.
- Software zur Behandlungsplanung: Tools zur Planung der Strahlentherapie.
- Laborausstattung:
- Blutgasanalysatoren und Hämatologieanalysatoren.
- Sterilisationsgeräte: Autoklaven, UV-Sterilisatoren.
- Medizinische Gase
- Geräte für die häusliche Gesundheitspflege:
- Überwachungsgeräte: Blutzuckermessgeräte und digitale Thermometer.
- Mobilitätshilfen: Gehhilfen, Elektroroller.
- Therapeutische Geräte (z. B. Dialysegeräte)
Lizenz zum Import von Medizinprodukten (MDIL)
Für den Import von Medizinprodukten ist ein gültiges MDMA erforderlich. Die SFDA kann jedoch bestimmte Medizinprodukte von der Registrierung ausnehmen und lediglich die Ausstellung einer Importlizenz für Medizinprodukte (MDIL) verlangen. Beispiele:
- Medizinische Geräte für Demonstrations- oder Schulungszwecke.
- Chemikalien (Fertigprodukte), unabhängig davon, ob sie als Medizinprodukt eingestuft sind oder zusammen mit Medizinprodukten verwendet werden (z. B. Gase zur Kalibrierung von Medizinprodukten sowie Chemikalien zur Sterilisation von Medizinprodukten, zur Herstellung von Prothesen und zur Konservierung von Gewebe oder Zellen). Ausgenommen hiervon sind die folgenden Produkte (sofern die Chemikalie als Medizinprodukt eingestuft ist und das Produkt MDMA enthalten muss).
- Radioaktive Stoffe
- Medizinische IVD
- Nicht-medizinische IVD
- Chemische Vorläufer.
- Die Destillationsapparate für Gesundheitsdienstleister oder Bildungseinrichtungen.
- Produkte für Forschungs- und Bildungszwecke.
- Halbfertige medizinische Geräte/Zubehör (sowie Roh- und Nicht-Rohchemikalien) für die lokale Fertigung (die Fertigung umfasst Aufarbeitung, Montage, Verpackung und Etikettierung).
Firmenregistrierung
Die SFDA reguliert auch Medizinproduktehersteller und legt dabei unterschiedliche Anforderungen für lokale und internationale Unternehmen fest. Nachfolgend sind die Anforderungen für beide Typen aufgeführt:
Lokales saudisches Unternehmen
Die SFDA lizenziert lokale Medizinprodukteunternehmen wie Importeure, Distributoren, Lagerhäuser, autorisierte Vertreter und Hersteller. Die lokale Unternehmenslizenz heißt Medical Device Establishment License (MDEL). Alle Unternehmen dieser Art müssen ein Qualitätsmanagementsystem (QMS) implementieren und über ein ISO 13485-Zertifikat verfügen.
Rechtlicher Hersteller
International zugelassene Hersteller müssen einen autorisierten Vertreter (AR) in Saudi-Arabien benennen; dies ist der erste Schritt zur Kommunikation mit der SFDA . Die Hauptverantwortung des AR liegt in der Sicherstellung der Produktkonformität vor und nach der Markteinführung. Der AR verwaltet den Registrierungsstatus, erleichtert den Versand, überwacht die Sicherheit, meldet Fälle und vertritt das Unternehmen in regulatorischen und rechtlichen Angelegenheiten.
Anmeldeservice
Unabhängig davon, ob Ihr Unternehmen neu auf dem saudischen Markt ist oder bereits dort aktiv ist, unterstützt Sie PharmaKnowl bei der Einhaltung der SFDA und erleichtert Ihr Geschäft, indem es Ihren Vertriebshändlern provisionsfrei Compliance, Einblicke und Versorgungsmanagement bietet.
PharmaKnowl ist ein SFDA lizenzierter Dienstleister in Saudi-Arabien und vertritt namhafte internationale Unternehmen. Kontaktieren Sie uns, um ein unverbindliches Gespräch zu vereinbaren oder weitere Informationen anzufordern.



