
Informazioni sull'autore

Mohammed Giobbe
Regulatory Consultant (Pharmacist BSc, Biomedical Informatics MSc). Con 19 anni di esperienza lavorativa presso SFDA, aziende multinazionali e come consulente professionista.
Per i produttori che desiderano entrare nel mercato saudita in rapida espansione, è essenziale orientarsi nella procedura di registrazione dei dispositivi medici SFDA in Arabia Saudita, in particolare nella procedura di autorizzazione all’immissione in commercio dei dispositivi medici (MDMA).
I requisiti relativi all’MDMA si sono evoluti per far fronte all’evoluzione degli standard internazionali, come l’evoluzione dalle Direttive UE sui Dispositivi Medici (MDD) ai Regolamenti sui Dispositivi Medici (MDR). Il continuo sviluppo delle normative sui dispositivi medici in Arabia Saudita ha aumentato la complessità di questo processo.
Ad esempio, anche i dispositivi medici a basso rischio (materiali di consumo e forniture) hanno iniziato a necessitare di un rappresentante autorizzato (AR) e dell’approvazione dell’MDMA.
Noi di PharmaKnowl offriamo supporto esperto per il processo di registrazione. Che si tratti di nuovi dispositivi o di modificare prodotti esistenti, il nostro team fornisce la visione strategica e la competenza tecnica necessarie per un ingresso tempestivo e di successo sul mercato saudita.
Questo articolo chiarisce i requisiti di registrazione dei dispositivi medici SFDA e definisce le differenze di classificazione.
Per altri tipi di registrazione del prodotto, fare riferimento alla pagina di registrazione SFDA .
Sommario
- Domande di registrazione
- Requisiti di registrazione
- Processo di registrazione
- Classificazione
- Raggruppamento di più dispositivi
- Linee temporali
- Commissioni
- Rinnovo
- Aggiornamento
- Certificato MDMA
- Categorie di prodotto
- Licenza di importazione di dispositivi medici (MDIL)
- Registrazione della società
- Servizio di registrazione
Domande di registrazione
La SFDA regolamenta i dispositivi medici in Arabia Saudita per garantirne sicurezza, efficacia e qualità. I produttori devono ottenere l’approvazione per l’MDMA prima di commercializzare i loro prodotti. Dopo l’approvazione, sono responsabili della segnalazione di sicurezza e di altre operazioni di gestione del ciclo di vita, come l’aggiornamento, il rinnovo e la presentazione dell’Identificativo Univoco del Dispositivo (UDI) .
Ecco una sintesi delle procedure di registrazione dei dispositivi medici:
MDNR (rischio basso) – Annullato
Le aziende registravano i dispositivi medici non sterili, non misurabili e a basso rischio tramite il Registro Nazionale dei Dispositivi Medici (MDNR), noto anche come “Elenco dei Dispositivi Medici”, che è esente da MDMA e AR. Tuttavia, a settembre 2022, la SFDA ha annullato la procedura MDNR . Pertanto, tutti i dispositivi devono disporre di un fascicolo tecnico per l’MDMA presso un AR designato in Arabia Saudita.
MDMA 1 (GHTF) – Annullato
In questa procedura di richiesta, la SFDA ha preso in considerazione le approvazioni dei paesi membri del GHTF, come Unione Europea, Stati Uniti, Canada, Australia e Giappone. Si è trattato di una procedura semplice da seguire, senza dover affrontare approfondite indagini tecniche. Tuttavia, entro la fine del 2021, la SFDA ha annullato questa procedura GHTF.
MDMA 2 (TFA)
Da gennaio 2022, tutti i dispositivi medici e gli IVD devono ottenere un’autorizzazione SFDA tramite la procedura di registrazione “Technical File Application” (TFA) (MDMA2). Questa procedura è il risultato dei continui aggiornamenti normativi dell’autorità, influenzati dai Regolamenti MDR e IVDR dell’UE. Richiede la presentazione della documentazione tecnica in modo chiaro, consultabile e organizzato.
Pertanto, i requisiti di registrazione sono diventati più rigorosi. È necessario soddisfare numerose condizioni e fornire studi specifici, come i rapporti di valutazione clinica (CER), i rapporti sui test di biocompatibilità e la necessità di condurre uno studio di follow-up clinico post-commercializzazione (PMCF) in Arabia Saudita.
Sebbene i requisiti TFA siano simili a quelli dell’UE (marchio CE), la SFDA non richiede un certificato CE. Tuttavia, se il fascicolo di prodotto non reca il marchio CE, verrà sottoposto a una rigorosa valutazione.
Requisiti di registrazione
Di seguito sono riepilogati i requisiti, che variano a seconda del tipo e della classe di prodotto. Ecco le sezioni principali del fascicolo di registrazione:
- Indice dei contenuti
- Descrizione del dispositivo
- Uso/scopo previsto
- Cronologia del dispositivo
- Classificazione dei dispositivi.
- Etichetta del dispositivo
- Modelli di dispositivi, accessori e varianti
- Istruzioni per l’uso
- Informazioni di progettazione
- Informazioni sulla produzione
- Principi essenziali (elenco EP) di sicurezza e prestazioni (elenco di controllo essenziale richiesto)
- Analisi rischi-benefici
- File di gestione del rischio (piano e rapporto)
- Test preclinici
- Rapporti di prova
- Rapporti sui test di biocompatibilità.
- Piano e relazione di indagine clinica
- Rapporto di valutazione clinica (CER)
- Follow-up clinico post-marketing (PMCF)
- Sorveglianza post-vendita (PMS), pianificazione e reportistica
- Rapporto periodico di aggiornamento sulla sicurezza (PSUR) per i dispositivi medici.
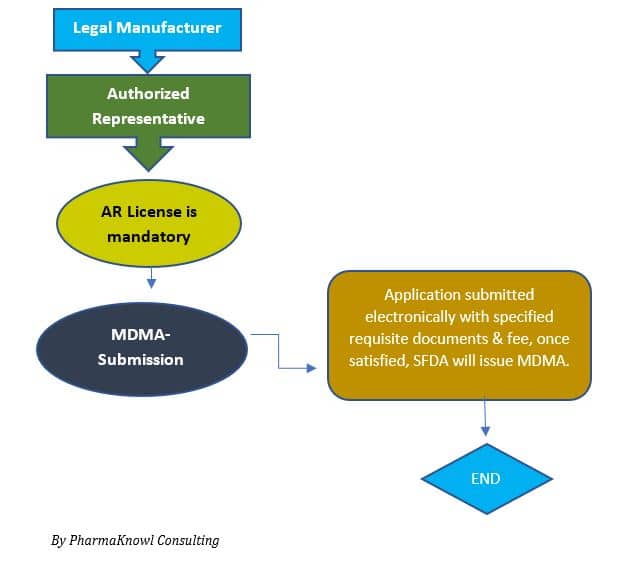
Processo di registrazione
Time needed: 90 days
Possiamo riassumere il processo di approvazione della registrazione del dispositivo medico (MDMA) nei seguenti pochi passaggi:
- Nomina di un rappresentante autorizzato (AR)
I produttori devono nominare un AR che rappresenti la loro azienda in Arabia Saudita.
- Analisi dei gap
Le aziende devono effettuare un’analisi approfondita delle lacune del dossier di registrazione rispetto a tutti i requisiti applicabili. Identificare le lacune è essenziale per determinare gli studi e i test pertinenti, come stabilità, CER, biocompatibilità, gestione del rischio e PMCF.
- Presentazione di MDMA
Il richiedente presenta il fascicolo tecnico alla SFDA .
- Validazione
SFDA garantisce le buone pratiche di invio senza valutarne il contenuto.
- Pagamento delle tasse
Il richiedente riceverà una fattura SFDA prima della valutazione.
- Valutazione dell’applicazione
I valutatori esamineranno il fascicolo e invieranno le loro richieste in più fasi.
- Approvazione MDMA
I produttori legali ricevono un certificato MDMA che consente loro di commercializzare il prodotto in Arabia Saudita.
Classificazione
I richiedenti devono classificare i loro prodotti secondo le norme di classificazione dei dispositivi medici SFDA nell’ambito della domanda di autorizzazione all’immissione in commercio di MDMA. Queste norme corrispondono al regolamento UE (certificato CE), in quanto rappresentano il riferimento non ufficiale SFDA , ma non necessariamente coincidono con esso.
Dispositivo medico
La classificazione dei dispositivi medici SFDA è di Classe A, B, C o D, in base alla loro classe di rischio. La classe è necessaria per determinare la procedura di registrazione e i relativi requisiti, come illustrato nella tabella seguente:
| Classificazione SFDA | Classe di rischio | Regola di classificazione MDR |
| Classe A | Basso | IO |
| Classe A – Sterile | Basso-medio | È |
| Classe A – Funzione di misura | Basso-medio | Io sono |
| Classe A – Strumenti chirurgici riutilizzabili | Basso-medio | Io sono |
| Classe B | Basso-medio | IIa |
| Classe C | Medio-alto | IIb |
| Classe D | Alto | III |
La classificazione avviene secondo quanto segue:
- Destinazione d’uso.
- Livello di rischio (probabilità e gravità del danno per i pazienti, gli utenti e altri).
- Invasività del corpo umano.
- Durata dell’utilizzo.
Dispositivi identici potrebbero essere classificati in modo diverso a seconda della parte del corpo interessata. Pertanto, l’uso previsto è fondamentale per determinare la corretta classificazione. Il riferimento all’uso previsto è riportato di seguito:
- Istruzioni per l’uso (IFU)
- Etichetta
- Materiali promozionali
- Scheda tecnica
Diagnostica in vitro (IVD)
Per quanto riguarda la diagnostica in vitro, la SFDA sta anche adottando il regolamento europeo sui dispositivi medici IVDR:
| Classificazione SFDA IVD | Classe di rischio | Regola di classificazione |
| Classe A | Basso rischio individuale e basso rischio per la salute pubblica | UN |
| Classe B | Rischio individuale moderato e/o basso rischio per la salute pubblica | B |
| Classe C | Rischio individuale elevato e/o rischio moderato per la salute pubblica | C |
| Classe D | Alto rischio individuale e alto rischio per la salute pubblica | D |
Il richiedente è responsabile della determinazione della classe del dispositivo medico-diagnostico in vitro mediante:
- Applicazione delle regole di classificazione per i dispositivi medici IVD e
- Considerando:
- Uso previsto
- Livello di rischio
Raggruppamento di più dispositivi
È consentito raggruppare più dispositivi nella stessa domanda; il richiedente può raggruppare fino a 50 articoli in un’unica domanda per l’MDMA. Tuttavia, i prodotti devono soddisfare diverse condizioni, come ad esempio la stessa classe di rischio e la stessa destinazione d’uso.
Linee temporali
Le tempistiche di valutazione variano in base a diversi fattori, tra cui la classe di rischio, la complessità del prodotto, il numero di prodotti inclusi nel bundle e la completezza del fascicolo. Lacune non identificate prima della presentazione rischiano di ritardare l’approvazione. Le tempistiche complete per il progetto di registrazione includono:
- Tempo di analisi dei gap
Questa parte dipende dal numero e dal tipo di lacune, dalla velocità del feedback del produttore e dall’esperienza del personale addetto alla regolamentazione. - Tempo di valutazione SFDA
Per le tempistiche ufficiali della revisione, fare riferimento al nostro articolo Cronologie SFDA .
Commissioni
Le tariffe per la domanda di autorizzazione all’immissione in commercio di MDMA variano in base alla classe di rischio del dispositivo medico e al numero di prodotti inclusi nella domanda. Per ulteriori informazioni, consultare il nostro articolo aggiornato sulle tariffe SFDA .
Rinnovo
La validità predefinita dell’MDMA è di tre anni. Quando la licenza è prossima alla scadenza, i produttori possono presentare una domanda di rinnovo per l’MDMA fino a tre mesi (90 giorni) prima della data di scadenza. Se si registra la licenza obsoleta MDMA1 (GHTF), la procedura di rinnovo sarebbe lunga e richiederebbe requisiti complessi, proprio come una nuova registrazione. Al contrario, il rinnovo per l’MDMA2 sarà più rapido.
Aggiornamento
Per apportare modifiche al dispositivo medico registrato, i produttori devono presentare una domanda di aggiornamento MDMA .
Certificato MDMA
SFDA rilascerà un certificato MDMA in arabo e in inglese contenente quanto segue:
- Informazioni sul produttore (titolare della licenza)
- Nome del dispositivo medico e informazioni sul gruppo di dispositivi medici.
- Periodo di validità
- Numero del certificato

Categorie di prodotto
Ecco alcuni esempi di tipologie e categorie di dispositivi medici in Arabia Saudita:
- Dispositivi diagnostici:
- Apparecchiature per la diagnostica per immagini: apparecchi a raggi X, scanner TC.
- Dispositivi di monitoraggio: elettrocardiografi e misuratori della pressione sanguigna.
- Dispositivi diagnostici in vitro: test di gravidanza, misuratori della glicemia.
- Dispositivi terapeutici:
- Apparecchi respiratori: ventilatori, nebulizzatori.
- Dispositivi di infusione: pompe per insulina e apparecchiature per terapia endovenosa.
- Dispositivi per la riabilitazione: sedie a rotelle, protesi.
- Dispositivi impiantabili:
- Dispositivi cardiovascolari: pacemaker, stent.
- Impianti ortopedici: protesi d’anca, barre spinali, sostituzioni articolari.
- Strumenti chirurgici:
- Strumenti da taglio: bisturi, forbici chirurgiche, pinze.
- Suturatrici e suture: suturatrici chirurgiche, suture assorbibili.
- Dispositivi dentali:
- Materiali restaurativi: corone dentali, otturazioni.
- Apparecchi ortodontici: apparecchi ortodontici, contenitori.
- Altro: trapani odontoiatrici
- Dispositivi oftalmici:
- Lenti correttive: lenti a contatto, occhiali.
- Attrezzature chirurgiche: apparecchi LASIK, lenti intraoculari.
- Software medico (SaMD) :
- Software diagnostico: intelligenza artificiale per l’analisi delle immagini.
- Software per la pianificazione del trattamento: strumenti per la pianificazione della radioterapia.
- Attrezzatura da laboratorio:
- AnalizzatoriAnalizzatori per analisi dei gas ematici e analizzatori per analisi ematologiche.
- Apparecchiature per la sterilizzazione: autoclavi, sterilizzatori UV.
- Gas medicali
- Dispositivi per l’assistenza sanitaria domiciliare:
- Dispositivi di monitoraggio: misuratori della glicemia e termometri digitali.
- Ausili per la mobilità: deambulatori, scooter elettrici.
- Dispositivi terapeutici (ad esempio, macchine per la dialisi)
Licenza di importazione di dispositivi medici (MDIL)
L’importazione di dispositivi medici richiede MDMA valida. Tuttavia, la SFDA potrebbe esentare alcuni dispositivi medici dalla registrazione e richiedere solo il rilascio di una licenza di importazione di dispositivi medici (MDIL). Ad esempio:
- Dispositivi medici a scopo dimostrativo o formativo.
- Prodotti chimici (prodotto finito), classificati come dispositivi medici o utilizzati insieme a dispositivi medici (ad esempio, gas utilizzati per la calibrazione dei dispositivi medici, nonché prodotti chimici utilizzati per garantire la sterilizzazione dei dispositivi medici, la produzione di protesi e la conservazione di tessuti o cellule). Sono esclusi i seguenti prodotti (se la sostanza chimica è classificata come dispositivo medico e il dispositivo deve contenere MDMA).
- Materiali radioattivi
- Diagnostica medica in vitro
- IVD non medico
- Precursori chimici.
- Apparecchi di distillazione per operatori sanitari o strutture educative.
- Prodotti per uso didattico e di ricerca.
- Dispositivi/forniture mediche semilavorati (e prodotti chimici grezzi e non grezzi) per la produzione locale (la produzione comprende la ristrutturazione, l’assemblaggio, il confezionamento e l’etichettatura).
Registrazione della società
La SFDA regolamenta anche le aziende produttrici di dispositivi medici, applicando requisiti diversi per le aziende locali e internazionali. Di seguito sono riportati i requisiti per entrambe le tipologie:
Azienda locale saudita
La SFDA rilascia le licenze alle aziende locali di dispositivi medici, come importatori, distributori, magazzini, rappresentanti autorizzati e produttori. La licenza aziendale locale è la Medical Device Establishment License (MDEL). Tutte le precedenti tipologie di aziende devono implementare un sistema di gestione della qualità (QMS) e possedere una certificazione ISO 13485 .
Produttore legale
I produttori internazionali autorizzati devono nominare un rappresentante autorizzato (AR) in Arabia Saudita; questo è il primo passo per consentire la comunicazione con l’ SFDA . La responsabilità principale dell’AR è la conformità del prodotto nelle attività pre/post-marketing. L’AR manterrà lo stato di registrazione, faciliterà le spedizioni, monitorerà la sicurezza, segnalerà i casi e rappresenterà l’azienda in questioni normative o legali.
Servizio di registrazione
Che la tua azienda sia nuova o già attiva nel mercato saudita, PharmaKnowl ti aiuterà a rispettare la SFDA e a semplificare la tua attività offrendo ai tuoi distributori servizi di conformità, approfondimenti e gestione della fornitura senza commissioni.
PharmaKnowl è un fornitore di servizi autorizzato SFDA in Arabia Saudita che rappresenta rinomate aziende internazionali. Contattateci per fissare un incontro conoscitivo o richiedere maggiori informazioni.



