
Informazioni sull'autore

Redattore normativo
Pubblicato dal team degli affari normativi di PharmaKnowl, ufficio di Riyadh.
La SFDA prevede rigorosi requisiti di etichettatura e imballaggio che le aziende devono rispettare durante la procedura di registrazione SFDA prodotti . Una volta approvate, le informazioni sull’etichettatura non devono essere modificate nemmeno minimamente per garantire la conformità del prodotto ed evitare il rifiuto della spedizione o il ritiro dal mercato.
In questo articolo verranno esaminati i requisiti di etichettatura per farmaci e dispositivi medici.
Etichettatura dei farmaci
I requisiti specifici di etichettatura SFDA devono essere applicati a tutti i componenti illustrativi del farmaco, inclusi astuccio, etichetta, lamina, foglietto illustrativo (Foglio Informativo per il Paziente) e Riassunto delle Caratteristiche del Prodotto (RCP). In questo documento, illustreremo i requisiti di alto livello per ciascuno dei componenti sopra menzionati, in quanto rappresentano uno dei principali requisiti di registrazione dei farmaci in Arabia Saudita .
Imballaggio esterno e interno (imballaggio secondario)
Nome del medicinale
La descrizione del prodotto deve essere visualizzata su più di tre facciate non opposte delle sei della scatola. Deve essere scritta in inglese e in arabo.
Dichiarazione della/e sostanza/e attiva/e
L’espressione del principio attivo deve essere presentata qualitativamente e quantitativamente per unità di dosaggio. Deve essere redatta in inglese e in arabo.
Dichiarazione di indicazione
L’indicazione del prodotto deve essere evidenziata sulla confezione, ad esempio per il trattamento di, la prevenzione di, ecc.
Elenco degli eccipienti
Gli eccipienti con effetti o azioni riconosciuti devono essere espressi qualitativamente. Tutti gli eccipienti devono essere indicati se il medicinale è somministrato per via parenterale, topica, oftalmica o inalatoria.
Forma farmaceutica e contenuto
Il contenuto deve essere indicato in peso, volume, numero di dosi o numero di unità di somministrazione (ad esempio, 10 compresse, 100 mL, ecc.).
Modalità e via di somministrazione
Il metodo di somministrazione e le istruzioni per un corretto utilizzo del medicinale devono essere chiaramente indicati, ad esempio “Agitare bene prima dell’uso”. Per le istruzioni d’uso, fare riferimento a (vedere il foglietto illustrativo allegato).
Avvertenza speciale
Sull’etichetta dovrebbe essere evidenziata un’avvertenza speciale, ad esempio: “Proteggere dalla luce solare, tenere fuori dalla vista e dalla portata dei bambini, solo per uso esterno”.
Condizioni di conservazione
È necessario specificare le condizioni di conservazione del prodotto, ad esempio conservare a temperatura inferiore a 25 ° C o conservare a temperatura inferiore a 30 ° C. Per i prodotti multidose è necessario aggiungere la stabilità in uso, ad esempio la durata di conservazione dopo la prima apertura deve essere di un mese, due mesi, tre mesi, ecc. Deve riflettere quanto affermato nello studio sulla stabilità SFDA .
Produttore e MAH
Devono essere menzionati il nome e l’indirizzo del produttore e del titolare dell’autorizzazione all’immissione in commercio.
Blister o strisce (Imballaggio primario)
Nome del medicinale
Il nome e il dosaggio del prodotto devono apparire su ogni alveolo del blister; se gli alveoli sono troppo piccoli, le informazioni devono essere ripetute in modo uniforme su tutta la striscia. Sono richieste in inglese e arabo, requisiti obbligatori per l’etichettatura SFDA di tutti i componenti.
Nome del titolare dell’autorizzazione all’immissione in commercio
È sufficiente solo il nome del marchio.
Date di produzione e scadenza
Le date devono essere espresse come, ad esempio, 02/2010 o febbraio 2010.
Numero di lotto
Il numero del lotto e la data di scadenza devono essere riportati alla fine di ogni blister.
Riepilogo delle caratteristiche del prodotto SPC
Sebbene l’RCP o il Riassunto delle Caratteristiche del Prodotto non siano stampati e allegati al farmaco, è necessario applicare i requisiti di etichettatura SFDA . Solo l’RCP approvato può essere distribuito ai medici e utilizzato per il materiale promozionale.
Informazioni primarie
Nome del medicinale, dosaggio e forma farmaceutica.
Composizione qualitativa e quantitativa
Il principio attivo deve essere espresso come dose per unità. È necessario includere una dichiarazione standard per gli eccipienti, ad esempio: “Per un elenco completo degli eccipienti, vedere la sezione ‘Elenco degli eccipienti'”.
Forma farmaceutica
È necessario indicare la forma farmaceutica del prodotto, ad esempio compressa rivestita con film, compressa a rilascio prolungato, ecc., insieme a una descrizione della forma farmaceutica. Se le compresse presentano una linea di frattura, è necessario specificare se facilita la rottura o se divide le compresse in dosi uguali.
Particolari clinici
Indicazioni terapeutiche
L’indicazione deve definire la malattia o la condizione target per il trattamento o la prevenzione e la sua indicazione negli adulti, nei neonati, nei lattanti, nei bambini e negli adolescenti di età (mesi, anni).
Posologia e modo di somministrazione
Le raccomandazioni posologiche devono essere specificate per intervallo di dose per ciascuna categoria, ove appropriato (determinare età/peso/superficie corporea di sottogruppi della popolazione, se necessario). Devono essere fornite informazioni su popolazioni speciali come pazienti anziani, con insufficienza renale, insufficienza epatica e popolazione pediatrica. Devono inoltre essere fornite misure preventive per la manipolazione e la somministrazione del prodotto.
Controindicazioni
Si tratta di casi in cui i medicinali non devono essere somministrati per motivi di sicurezza, vale a dire in caso di ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti.
Avvertenze speciali e precauzioni d’impiego
Informazioni sui rischi specifici, misure specifiche di minimizzazione del rischio, reazioni avverse, informazioni sulla sicurezza,
Interazioni
Studio di interazione clinica (in vivo), interazione farmacodinamica, interazione farmacocinetica.
Fertilità, gravidanza e allattamento
Le raccomandazioni per l’uso nelle donne in gravidanza, in allattamento e in età fertile devono essere basate sugli studi clinici , sugli studi non clinici e sull’attività farmacologica.
Effetti sulla capacità di guidare veicoli e sull’uso di macchinari
Effetti indesiderati
Riepilogo del profilo di sicurezza, reazione avversa e indirizzo dell’autorità nazionale a cui segnalare eventuali effetti collaterali.
Overdose
Effetto di diversi livelli di dosaggio assunti accidentalmente, per errore o in caso di tentativi di suicidio.
Proprietà farmacologiche
Il contenuto deve essere indicato in peso, volume, numero di dosi o numero di unità di somministrazione del medicinale (ad esempio, 10 compresse, 100 mL, ecc.).
Particolari farmaceutici
- Elenco degli eccipienti
- Incompatibilità
- Durata di conservazione
- Precauzioni speciali per la conservazione
- Natura e contenuto del contenitore.
Precauzioni speciali per lo smaltimento e la manipolazione
Requisiti speciali per lo smaltimento, se non specificato diversamente, è possibile indicare “nessun requisito speciale”. Lo smaltimento deve essere effettuato in conformità alle normative locali.
Titolare dell’autorizzazione all’immissione in commercio
Numero di autorizzazione all’immissione in commercio
Data della prima autorizzazione/rinnovo dell’autorizzazione
Data di revisione del testo
Foglio informativo per il paziente (PIL)
Che cosa è (nome di fantasia) e a cosa serve
Nome di fantasia, concentrazione, forma farmaceutica, principio attivo, indicazione terapeutica e beneficio dell’uso di questo medicinale.
Prima di assumere o utilizzare il prodotto
Controindicazioni, opportune precauzioni d’impiego/avvertenze speciali, interazioni con altri medicinali/cibi/bevande, informazioni per l’uso in donne in gravidanza o in allattamento, informazioni sulla fertilità, guida di veicoli e uso di macchinari, avvertenze sugli eccipienti.
Come assumere o utilizzare il prodotto
Dosaggio, Modalità e via di somministrazione, Frequenza di somministrazione, Istruzioni per un uso corretto, Durata del trattamento, Sovradosaggio, Uso irregolare del medicinale,
Possibili effetti collaterali
Descrizione degli effetti collaterali, ulteriori effetti collaterali nei bambini e negli adolescenti.
Come memorizzare il nome del prodotto
Durante lo stoccaggio e le condizioni di conservazione sono necessarie particolari indicazioni e precauzioni.
Ulteriori informazioni
Informazioni sul/i principio/i attivo/i ed eccipiente/i, forma farmaceutica, natura e contenuto del contenitore, nome e indirizzo del titolare dell’autorizzazione all’immissione in commercio e del produttore responsabile del rilascio dei lotti, data di revisione del foglio illustrativo (FI) e indirizzo dell’autorità nazionale per la segnalazione di eventuali effetti indesiderati.
Informazioni sul titolare dell’autorizzazione all’immissione in commercio
Nome e indirizzo
Informazioni del produttore
Nome e indirizzo
Questo foglio illustrativo è stato rivisto l’ultima volta il
Data: mese e anno
Per segnalare eventuali effetti collaterali
Aggiungere in questa sezione tutte le informazioni sulla farmacovigilanza e sulla QPPV e le istruzioni su come segnalare.
Etichettatura dei dispositivi medici
I requisiti di etichettatura SFDA per i dispositivi medici si applicano ai materiali di imballaggio, alle etichette e alle istruzioni per l’uso (IFU) e devono essere presentate di conseguenza nelle domande di registrazione dei dispositivi medici (MDMA). Si applicano sia ai dispositivi a basso rischio e dispositivi medici ad alto rischio.
Il termine etichettatura è un termine collettivo che comprende:
- L’etichetta,
- Istruzioni per l’uso (talvolta denominate manuale dell’operatore) e
- Qualsiasi altra informazione relativa all’identificazione, alla descrizione tecnica, allo scopo previsto e all’uso corretto del dispositivo medico,
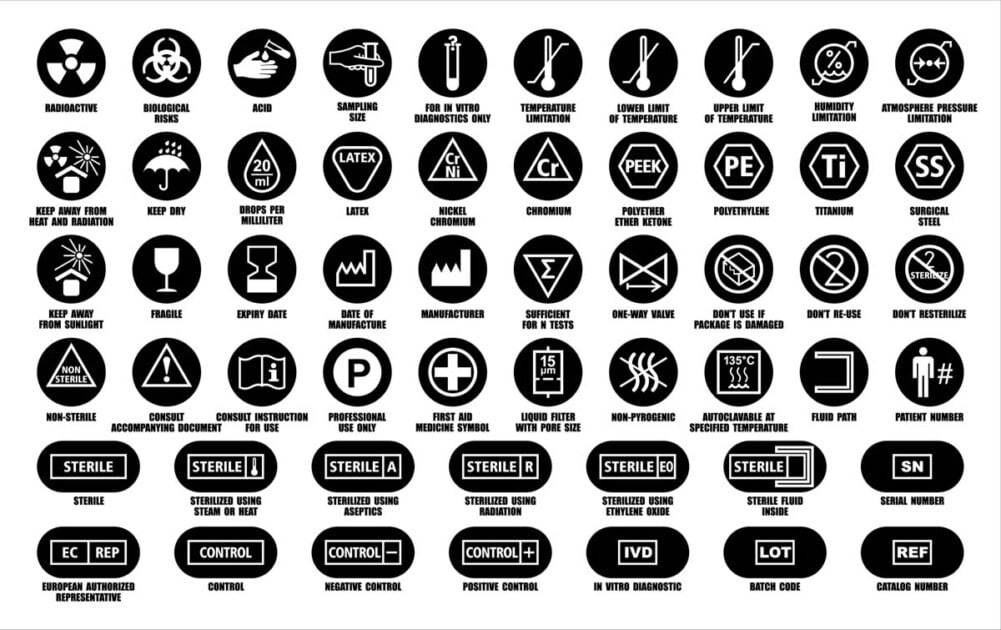
Il termine etichetta descrive informazioni scritte, stampate o grafiche che sono:
- Applicato o presente sul dispositivo medico stesso (incluso il display elettronico)
- Sulla confezione di:
- Ogni unità (involucro)
- Dispositivi multipli (contenitori)
- Su un foglietto illustrativo (viene utilizzato quando è poco pratico o inappropriato apporre un’etichetta direttamente sul dispositivo medico stesso. Poco pratico significa quando vincoli fisici impediscono che ciò accada).
Lo scopo principale dell’etichettatura è:
- Identificare il dispositivo medico e il suo produttore,
- Descrivere l’uso previsto e le prestazioni del dispositivo,
- Descrivere come il dispositivo deve essere utilizzato, mantenuto e conservato, e
- Fornire informazioni su eventuali rischi residui, avvertenze, limitazioni o controindicazioni del dispositivo.
Istruzioni per l’uso (IFU)
Con il termine “Istruzioni per l’uso (IFU)” si intendono le informazioni fornite dal produttore per informare l’utilizzatore del dispositivo:
- Della destinazione d’uso e dell’uso corretto del dispositivo medico.
- Di eventuali precauzioni da adottare.
Le istruzioni per l’uso potrebbero non essere necessarie o essere abbreviate per i dispositivi se questi possono essere utilizzati in modo sicuro e come previsto dal produttore senza tali istruzioni per l’uso (ad esempio, lenti ottiche, bastoni da passeggio o semplici medicazioni per ferite).
Linguaggio di etichettatura
Quando l’utilizzatore del dispositivo medico è:
- Probabilmente si tratta di personale professionalmente qualificato e l’etichettatura dovrà essere in inglese.
- Per i non addetti ai lavori, l’etichetta e le istruzioni per l’uso devono essere redatte in arabo e in inglese, ove possibile. Ove ciò non sia possibile, la lingua utilizzata nell’etichetta e nelle istruzioni per l’uso sarà l’arabo.
In entrambe le situazioni, il testo deve essere redatto in termini facilmente comprensibili per l’utente previsto, commisurato alla sua conoscenza tecnica, esperienza, istruzione o formazione.
Lingua delle istruzioni del produttore
Le istruzioni per la manipolazione, la conservazione, il trasporto, l’installazione, la manutenzione e lo smaltimento dei dispositivi medici devono essere redatte in inglese e, ove giustificato, in arabo. Il testo deve essere redatto in termini facilmente comprensibili per l’utilizzatore previsto, commisurato alle sue conoscenze tecniche, esperienza, istruzione o formazione, laddove persone senza qualifica medica possano svolgere tale attività. Qualora il dispositivo sia progettato per un pubblico non specializzato, le istruzioni per la manipolazione, la conservazione, il trasporto e la manutenzione dei dispositivi medici devono essere redatte in arabo e in inglese.
Mezzo di etichettatura
Le etichette devono essere fornite in un formato leggibile dall’uomo, ma possono essere integrate da formati leggibili da una macchina, come l’identificazione a radiofrequenza (RFID) o i codici a barre.
Contenuto dell’etichettatura
Nome del dispositivo
Il nome commerciale o il marchio del prodotto devono essere stampati sull’etichetta. Analogamente, sull’etichetta deve essere stampato il numero di modello del prodotto.
Nome e indirizzo del produttore
Il nome e l’indirizzo del produttore devono essere stampati sulle etichette e devono corrispondere lettera per lettera ai dati di indirizzo indicati nel contratto di autorizzazione all’immissione in commercio presentato e nella successiva licenza di autorizzazione all’immissione in commercio. Qualsiasi discrepanza nei dati di indirizzo comporterebbe il ritorno della domanda presentata alla SFDA , creando quindi complicazioni nell’ottenimento dell’autorizzazione all’immissione in commercio per dispositivi medici.
Produttore legale nei casi OEM/OBL
OEM è l’abbreviazione di Original Equipment Manufacturer, mentre OBL è Own Brand Labelling.
Nei casi OEM/OBL, in cui la giurisdizione UE è stata scelta come base per la domanda di autorizzazione all’immissione in commercio di MDMA, SFDA considera come produttore legittimo il nome del produttore accanto al “Simbolo del produttore” sull’etichetta. In caso contrario, è necessario presentare alla SFDA i seguenti documenti:
- Copia dei certificati di valutazione della conformità basati sul percorso di valutazione della conformità utilizzato (ad esempio, certificato CE, certificato DE, certificato di garanzia della qualità della produzione, ecc.)
- Copia della Dichiarazione di Conformità dei prodotti.
- Copia dell’etichettatura (incluse le istruzioni per l’uso)
- Rapporto di audit più recente (se applicabile)
Alimentazione elettrica
Nel caso in cui l’apparecchio sia collegato ad una rete elettrica a corrente alternata, indicazione della frequenza nominale (60 Hertz) e dei valori di tensione con le relative tolleranze per i quali il prodotto è stato progettato.
Etichettatura IVD
Ciò indica che il prodotto è destinato all’uso diagnostico in vitro se si tratta di un dispositivo medico IVD.
Condizioni di conservazione
Se del caso, indicazione delle eventuali condizioni speciali di conservazione e manipolazione applicabili.
Avvertenze o precauzioni
Eventuali avvertenze, precauzioni, limitazioni o controindicazioni.
Numero di lotto o di lotto
Il codice batch/numero di LOTTO o il numero di serie consente di applicare azioni di conformità post-commercializzazione se è necessario tracciare o richiamare il prodotto. Tuttavia, per gli accessori dei dispositivi medici IVD, questo può essere sostituito con un numero di controllo e per il software, deve essere sostituito con un numero di versione.
Data di scadenza
Un’indicazione univoca della data fino alla quale il prodotto può essere utilizzato in sicurezza (ad esempio su dispositivi sterili o monouso), ove pertinente.
Quantità
Ove pertinente, l’indicazione della quantità netta del contenuto è espressa in termini di peso o volume, conteggio numerico o qualsiasi combinazione di questi o altri termini che riflettano accuratamente il contenuto della confezione.
Monouso o multiuso
Se il prodotto è monouso, un’indicazione in tal senso.



