
The SFDA has strict labelling and packaging requirements that companies must comply with during the product registration process. Once approved, the labelling information must not be altered even minimally to maintain product compliance and avoid shipment rejection or withdrawal from the market.
This article will review the labelling requirements for drugs and medical devices.
Table of contents
Drug Labelling
Specific SFDA labelling requirements must be applied to all the drug artwork components, including carton, label, foil, leaflet insert (patient information leaflet-PIL), and the Summary of Product Characteristics SPC. We will outline the high-level requirements for each of the abovementioned components here, as they are one of the leading drug registration requirements in Saudi Arabia.
Outer and Inner Packaging (Secondary Packaging)
Name of the medicinal product
The product description is to be displayed on more than three non-opposing faces of the box’s six faces. It should be written in English and Arabic.
Statement of the active substance(s)
The active substance’s expression should be presented qualitatively and quantitatively per dosage unit. It should be written in English and Arabic.
Statement of Indication
The product indication should be highlighted on the pack. e.g., for the treatment of, the prevention of, etc
List of excipients
Excipients with recognised effects or actions should be expressed qualitatively. All excipients must be stated if the medicinal product is parenteral, topical, eye preparation, or used for inhalation.
Pharmaceutical form and contents
The contents should be mentioned by weight, volume, number of doses, or number of administration units (e.g., 10 tablets, 100 mL, etc.).
Method and route(s) of administration
The method of administration and directions for proper medicinal product use should be clearly stated, e.g., “Shake well before use. For the direction of use, it can be referred to (see enclosed leaflet)
Special warning
A special warning should be highlighted on the label, e.g., “Protect from sunlight, Keep out of sight and reach of children, for external use only. “
Storage condition
The product storage condition should be mentioned, e.g., Store below 250 °C or Store below 30 °C. In-use stability should be added for multidose products, e.g., shelf life after the first opening should be one month, two months, three months, etc. It has to reflect what is stated in the SFDA stability study.
Manufacturer and MAH
The manufacturer and Marketing Authorization Holder’s Name and Address should be mentioned.
Blisters or strips (Primary Packaging)
Name of the medicinal product
The name and strength of the product should appear over each blister pocket; if the pockets are too small, the information should be repeated in a pattern across the entire strip. It is required in English and Arabic, which are mandatory SFDA labelling requirements for all parts.
Name of the marketing authorisation holder
Brand name only is sufficient.
Manufacturing and Expiry dates
Dates should be expressed as, e.g., 02/2010 or Feb 2010.
Batch number
The batch (lot) number and expiry date should be at the end of each blister strip.
Summary of Product Characteristics SPC
Although the SPC or SmPC is not printed and enclosed with the drug product, the SFDA labelling requirements must be applied. Only the approved SmPC can be circulated to the practitioners and used for promotional materials.
Primary Information
Name of the medicinal product, strength, and pharmaceutical form.
Qualitative and Quantitative composition
The active substance should be expressed as a dose per unit. A standard statement should be included for excipients, e.g., ‘For a complete list of excipients, see the section List of Excipients.
Pharmaceutical form
The product dosage form should be mentioned, e.g., Film-coated tablet, sustained-release tablet, etc., along with a description of the dosage form. If the tablets are designed with a scoreline, information should be given on whether it facilitates breaking or divides the pills into equal doses.
Clinical particulars
Therapeutic Indications
The indication should define the target disease or condition for treatment or prevention, as well as its indication in adults, neonates, infants, children, and adolescents of various ages (in months or years).
Posology and method of administration
Dose recommendations should be specified per dose interval for each category where appropriate (determine age/weight/body surface area of subsets of the population as proper). Information on special populations like elderly patients, renal impairment, hepatic impairment, and the pediatric population should be provided. Also, preventive measures for handling and administering the product should be provided.
Contraindications
These are cases where medicinal products must not be administered due to safety concerns, i.e., hypersensitivity to the active substance or any of the excipients.
Special warning and precautions for use
Information on specific risks, specific risk minimisation measures, adverse reactions, safety information,
Interactions
Clinical interaction study (in vivo), Pharmacodynamic interaction, Pharmacokinetic interaction.
Fertility, Pregnancy, and Lactation
Recommendations for use in pregnant and lactating women and women of childbearing potential should be based on clinical trials, non-clinical studies, and pharmacological activity.
Effects on the ability to drive and use machines
Undesirable effects
Summary of the safety profile, adverse reactions, and the national authority address for reporting any side effects.
Overdose
Effect of different dosing levels taken accidentally, by mistake or by suicide attempts.
Pharmacological properties
The contents should be specified by weight, volume, number of doses, or number of units of medicinal product administration (e.g., 10 tablets, 100 mL).
Pharmaceutical particulars
- List of excipients
- Incompatibilities
- Shelf life
- Special precautions for storage
- Nature and contents of the container.
Special precautions for disposal and handling
Special requirements for disposal can be specified; otherwise, “no special requirements” can be mentioned. The disposal should be according to local requirements.
Marketing Authorization Holder
Marketing Authorisation Number
Date of first Authorisation/Renewal of the Authorization
Date of revision of the text
Patient Information Leaflet (PIL)
What (invented name) is, and what is it used for
The invented name, strength, pharmaceutical form, active ingredient, therapeutic indication, and benefit of using this medicine.
Before you take or use the product
Contraindication, appropriate precautions for use/special warnings, interactions with other medicines/food/drinks, information for use in pregnant or breastfeeding women, information on fertility, driving and using machines, and excipient’s warnings.
How to take or use the product
Dosage, Method and route(s) of administration, frequency of administration, Instructions for proper use, Duration of treatment, overdose, irregular use of medicine,
Possible side effects
Description of side effects, additional side effects in children and adolescents.
How to store the product name
Particular directions and precautions are required during storage and storage conditions.
Further information
Active substance(s) and excipient(s) information, Pharmaceutical form, nature and contents of container, name and address of the marketing authorisation holder and the manufacturer responsible for batch release, the revision date of the PIL, and the National authority address for reporting any side effect.
Marketing Authorization Holder Information
Name and Address
Manufacturer information
Name and Address
This leaflet was last revised on
Date: Month and Year
To report any side effects
To add under this section all the pharmacovigilance and QPPV information and instructions on how to report.
Medical Devices Labelling
The SFDA labelling requirements for medical devices apply to the packaging materials, labels, and instructions for use (IFU) and must be submitted accordingly in the medical device registration applications (MDMA). They apply to both low-risk and high-risk medical devices.
The term labelling is a collective term comprising:
- The label,
- Instruction for use (sometimes referred to as the operator’s manual), and
- Any other information that is related to identification, technical description, intended purpose, and proper use of the medical device,
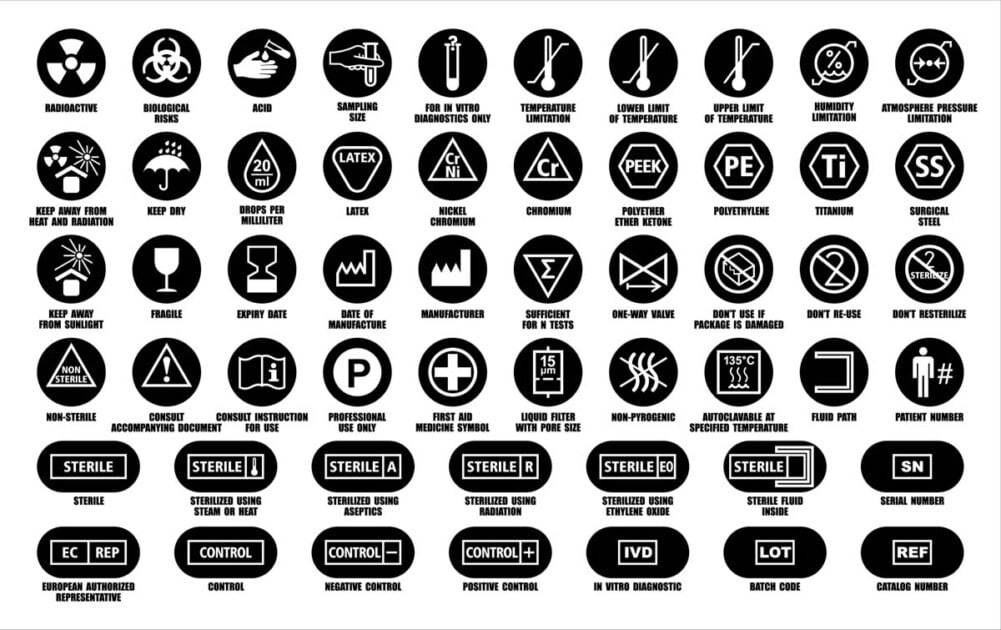
The term label describes written, printed, or graphic information that is:
- Affixed to or appearing on the medical device itself (including electronic display)
- On the packaging of:
- Each unit (wrapper)
- Multiple devices (containers)
- On a package insert (is used where it is impractical or inappropriate to affix a label directly on the medical device itself. Impractical means where physical constraints prevent this from happening).
The primary purpose of labelling is to:
- Identify the medical device and its manufacturer,
- Describe the device’s intended use and performance,
- Describe how the device should be used, maintained, and stored, and
- Provide information on any residual device risks, warnings, limitations, or contraindications.
Instructions For Use (IFU)
The term “Instructions for Use (IFU)” means the information provided by the manufacturer to inform the device user:
- Of the medical device’s intended purpose and proper use.
- What are any precautions to be taken?
IFU may not be needed or may be abbreviated for devices if they can be used safely and as intended by the manufacturer without any such Instructions For Use (e.g., Optical lenses, walking sticks, or simple wound dressings).
Labeling Language
Where the user of the medical device is:
- Likely to be professionally qualified, then the labelling shall be in English.
- Layperson, then the Label and IFU shall be in Arabic and English wherever feasible. Where this is not feasible, the language used in the label and IFU shall be Arabic.
In both situations, the text shall be written in terms readily understood by the intended user, commensurate with their technical knowledge, experience, education, or training.
Language of Manufacturer’s Instructions
Instructions for handling, storage, transportation, installation, maintenance, and disposal of the medical devices shall be in English and, where justified, in Arabic. The text shall be written in terms readily understood by the intended user, commensurate with their technical knowledge, experience, education, or training, where persons without medical qualifications may undertake such work. Where the device is designed for laypersons, instructions for handling, storing, transporting, and maintaining the medical device shall be provided in both Arabic and English.
Medium of Labelling
Labels shall be provided in a human-readable format but may be supplemented by machine-readable forms, such as radio-frequency identification (RFID) or bar codes.
Content of Labelling
Name of device
The product trade or brand name should be printed on the label. Likewise, the product Model No is printed on the label.
Name and address of the manufacturer
The manufacturer’s name and address must be printed on labels, and they must match exactly with the address details specified in the submitted AR agreement and subsequent AR license. Any discrepancy in address details would result in the return of the submitted application to SFDA and thus create complications in acquiring medical device marketing authorisation.
Legal Manufacturer in OEM /OBL Cases
OEM is the abbreviation of Original Equipment Manufacturer, while OBL is Own Brand Labelling.
The legal manufacturer in OEM / OBL cases, where the EU jurisdiction has been selected as the basis of the MDMA application, SFDA considers the manufacturer’s name next to the “Manufacturer Symbol” on the labelling as the legal manufacturer. If this is not applicable, the following documents should be submitted to SFDA:
- Copy of conformity assessment certificates based on the used conformity assessment route (i.e., CE certificate, DE certificate, Production Quality Assurance Certificate, etc.)
- Copy of products” Declaration of Conformity.
- Copy of labelling (including the Instruction for Use)
- Most recent Audit report (if applicable)
Power Supply
When the device is connected to an AC power supply, an indication of the nominal frequency (60 Hz) and the voltage values with their tolerances for which the product has been designed.
IVD Labeling
This indicates that the product is for in vitro diagnostic use if it is an IVD medical device.
Storage Conditions
Where applicable, an indication of any special storage and handling conditions that apply.
Warning or Precautions
Any warnings, precautions, limitations, or contraindications.
Batch or Lot Number
The batch code/ LOT number, or serial number, allows for post-market compliance actions to be applied if there is a need to trace or recall the product. However, for accessories of IVD medical devices, this may be substituted with a control number, and for software, it shall be replaced with a version number.
Expiry date
An unambiguous indication of the date until when the product may be used safely (e.g., on sterile or single-use disposables) where this is relevant.
Quantity
Where relevant, the net quantity of contents is expressed in terms of weight, volume, numerical count, or any combination of these, or other terms that accurately reflect the package contents.
Single or multiple-use
If the product is intended for single use, an indication of this fact should be provided.
About the Author

Contributed by the PharmaKnowl regulatory affairs team, based in Riyadh. Written and reviewed by our SFDA-experienced consultants.
Resources




Services
Events


