
The drug sector within the Saudi Food and Drug Authority (SFDA) regulates pharmaceutical and biotechnology products in Saudi Arabia, setting drug registration requirements and post-marketing obligations. These requirements have evolved significantly since 2009, the year the SFDA began operations, when it adopted the International Council for Harmonization (ICH) guidelines and improved its review and approval processes.
In the Marketing Authorization Application (MAA), the required drug dossier progressed gradually from paper-based to the electronic Common Technical Document (eCTD) format, passing through interim formats such as early CTD and the Non-eCTD electronic Submissions (NeeS).
In this post, we outline the SFDA drug registration requirements, how to prepare the drug file for different application types, and the various stages of the registration process.
For products other than medicines, you may refer to the Products Registration article.
Key Takeaways
- The SFDA regulates pharmaceuticals, biosimilars, and biotechnology medicines in Saudi Arabia
- Key drug registration requirements include having a local agent or consulting partner, a compliant dossier, and studies conducted in accordance with SFDA requirements, such as stability and bioequivalence.
- The authority offers several application types and accelerated pathways
- The authority incentivizes the registration of drugs in short supply.
Table of contents
- How to Plan for Drug Submission?
- Who can apply to the SFDA?
- Drug Registration Requirements
- Guidelines
- Drug Dossier Requirements
- Drug Application
- Submission
- Validation
- Approval Timelines
- Review and Approval Process
- Accelerated Review and Designations
- Life Cycle Management (LCM)
- Variation
- How PharmaKnowl Supports Registration Projects
How to Plan for Drug Submission?
Developing a regulatory strategy before initiating the registration project is a best practice when preparing for an SFDA drug submission. This strategy is essential for making informed business decisions and ensuring a seamless registration process. It provides the necessary visibility into the regulatory impact ahead for all functions.
Having a strategy should not be considered a luxury, even for well-established markets. Since the registration requirements are heavily considered in the strategy, the project will be enhanced, and progress will be smooth, without pitfalls.
Read more: How to Develop a Regulatory Strategy for the Saudi Market.
Who can apply to the SFDA?
Multinational companies can apply to the SFDA in different pathways, whether directly or indirectly. Essentially, the international Marketing Authorization Holder (MAH) will be the holder of the Marketing Authorization (MA) in Saudi Arabia for any drug registered in the SFDA.
Local regulations permit specific legal entities to submit applications to the Saudi FDA. The following are the available options for local and international companies.
Local Companies
Local Saudi companies can submit their applications directly to the SFDA, provided they hold the appropriate licenses for the relevant activity and have access to the relevant electronic systems.
International Companies
International companies can submit to the SFDA through one of the following entities:
Legal Entity (Independent SFDA Account)
To establish a local legal entity in Saudi Arabia as a foreign investment firm through the Ministry of Investment (MISA). This option could be feasible for multinational companies with well-established experience in the Saudi market.
Scientific Office (Independent SFDA Account)
International companies can establish a scientific office to enable direct submissions to the SFDA.
Local Distributors (Shared SFDA Account)
Local distributors can apply on behalf of the international companies. While it is cost-effective, it often falls short in critical areas such as confidentiality, regulatory expertise, dedication, and strategic alignment. All of these factors might lead to conflicts of interest, longer timelines, and a complex regulatory journey.
Consulting Company (Independent SFDA Account)
The SFDA allows international Pharma, Biotech, and MedTech companies to register new products in their own SFDA electronic accounts independently while continuing to conduct commercial operations through their local distributors. Here are a few essential points about this option:
- Creation: SFDA-approved local consulting firms can establish the international SFDA account to split regulatory functions from distributors.
- Process: Under this account, the global company can register new products and transfer their approved applications from the distributors’ accounts to its own while keeping the distributors appointed.
- Obligation: This approach does not oblige the global company to establish a legal presence in the market.
- Benefits: In return, it offers a range of benefits over reliance on distributor regulatory support, including:
- Independent: A dedicated private SFDA electronic account.
- Professional Regulatory: Utilize experienced consultants to work on the applications and officially represent the international company to help navigate complex regulations.
- SFDA Communication: High-level SFDA accessibility and professional engagement.
- Control and Oversight: Retain power over licenses and the ability to change appointed distributors.
- Confidentiality: Maintain strict data privacy and confidentiality of proprietary information.
- Accelerated Market Entry: Enables faster submissions to the SFDA while searching for and negotiating with potential commercial partners in a stronger position.
To learn more about all the entry options, you may contact us or request an online exploratory meeting.
Drug Registration Requirements
The most common question we receive is, “What are the SFDA registration requirements?” This question is tricky to answer with a confined checklist, since each required document has specific compliance measures based on the drug type, not to mention the comprehensive due diligence that extends beyond the drug file to the entities and facilities behind it.
Therefore, the information provided here enables the regulatory professionals to discover the depth and length of the requirements. In this direction, I segmented such interconnected processes into four sections:
- Applicable SFDA Guidelines
- Drug Dossier Structure
- Drug Application Types
- eCTD Submission
Guidelines
According to the drug type, you will find below the related SFDA drug registration guidelines:
Human Drug
- Data Requirements for Human Drugs Submission
- Guidance for Submission
- Guidance for priority review
- Stability Guidelines
- Drug Master File (DMF) guidelines
- Summary of Product Characteristics (SPC), Patient Information Leaflet (PIL), and Labeling requirements
- The naming of Medicinal Products
- Tamper-Evident Packaging Guidelines
- Graphic Design of Medication Packaging
- Registration According to Verification and Abridged
- Module 1 Specifications Guidelines for Bioequivalence
- Biosimilars Guidelines
- Investigational New Drugs (IND) Requirements
- Pharmaceutical Reference Standards
- Biowaiver Guidelines
- Production and Quality Control of Vaccines
- CAR-T Cell Product Registration
- Advanced Therapy Medicinal Products (ATMPs)
- Literature-Based Submissions in SFDA
Regarding generic drugs, the SFDA safeguards the patent rights and Intellectual Property (IP) of innovator products through a clear framework for patented drugs. This includes the requirement to obtain a freedom-to-operate (FTO) letter and to meet other patent-related conditions before submitting a generic drug application.
Herbal Drug
- Data Requirements for Herbal & Health Products Submission
- Guidance for Presenting PIL and Labeling Information on Herbal and Health Products
- General Rules For Products Containing Vitamins And Minerals
Veterinary Drug
- Veterinary Drug Registration in SFDA
- Veterinary Drug Residues
- Veterinary Herbal Drug Registration in KSA
- SFDA VNeeS specifications for the veterinary medicinal product
- Data Requirements for Veterinary Medicinal Products
- SFDA SPC, Leaflet, and Labeling for Veterinary Products
Drug Dossier Requirements
Dossier Files
- New drug
These are the innovative drugs equivalent to NDAs, including biosimilars.- All five modules are required.
- Generic drug
- M1: All sections
- M2: 2.1, 2.2, 2.3, 2.5.2
- M3: All sections.
- M4: NA
- M5: Only 5.1, 5.2, 5.3.1.2, 5.3.1.3, 5.3.1.4, 5.3.7, and 5.4
- Health and Herbal Products
- M1: All sections.
- M3: All sections.
- M5: All sections
- Veterinary Drug:
- Part 1
- Part 2
- Part 3
- Part 4
Download the Full Checklist: SFDA Drug Dossier Requirements.
Dossier Format
- Human drug: eCTD.
- Herbal and health products: CTD
- Veterinary drug: vNEES or CTD.
Module 1 Requirements
- 1.0 Cover letter
- 1.1 Comprehensive table of contents
- 1.2 Application Form
- 1.3 Product Information
- 1.3.1 SPC
- 1.3.2 Labeling
- 1.3.3 PIL
- 1.3.3.1 Arabic leaflet
- 1.3.3.2 English leaflet
- 1.3.4 Artwork (Mock-ups)
- 1.3.5 Samples
- 1.4 Information on the experts
- 1.4.1 Quality
- 1.4.2 Non-clinical
- 1.4.3 Clinical
- 1.5 Environmental Risk Assessment
- 1.5.1 Non-Genetically Modified Organism (Non-GMO)
- 1.5.2 GMO
- 1.6 Pharmacovigilance
- 1.6.1 Pharmacovigilance System
- 1.6.2 Risk Management Plan
- 1.7 Certificates and Documents
- 1.7.1 GMP Certificate
- 1.7.2 CPP or Free-sales
- 1.7.3 Certificate of Analysis – Drug Substance / Finished Product
- 1.7.4 Certificate of Analysis – Excipients
- 1.7.5 Alcohol-content declaration
- 1.7.6 Pork- content declaration
- 1.7.7 Certificate of Suitability for Transmissible Spongiform Encephalopathies (TSE)
- 1.7.8 The diluents and coloring agents in the product formula
- 1.7.9 Patent Information
- 1.7.10 Letter of access or acknowledgment to DMF
- 1.8 Pricing
- 1.8.1 Price list
- 1.8.2 Other related documents
- 1.9 Responses to questions
Drug Application
Applicants must complete the drug registration application and submit it via the Saudi Drug Registration (eSDR) system. It is a web portal available for local Saudi companies; it enables the applicants to do the following:
- Fill out and export the module 1 application.
- Pay the application fees.
- Submit the dossier.
- Receive SFDA Inquiries (RFI).
- Receive SFDA decisions.
- Print the registration certificate.
- Submit variations & renewals (life cycle management).
Application Types
Drug applications in eSDR come in three major types with more subtypes, as follows:
- Human Medicinal Product
- New Drug
- Biological
- Radiopharmaceutical Drug
- Generic (Multisource) Drug
- Health / Herbal Products
- Veterinary Product
- New Drug
- Biological Drug
- Generic (Multisource) Drug
- Herbal Product
- Health product
Application Fees
There is a matrix of fees related to drug registration, which we detailed in our post: SFDA Fees.
Submission
The following image shows the application submission process, which involves two layers of validation.
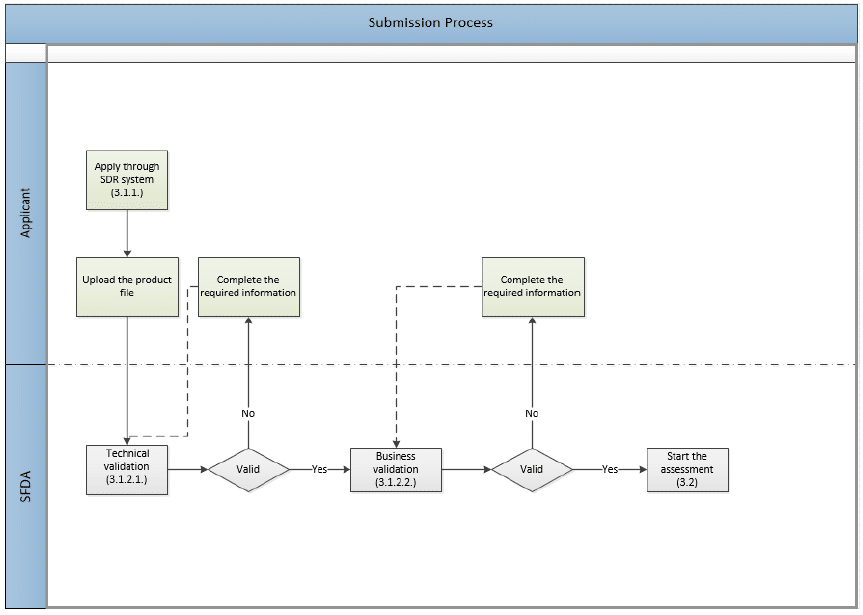
Validation
Technical Validation
The SFDA receives the submitted dossier and electronically validates the compiled eCTD in accordance with the SFDA eCTD Validation Criteria. Read: What are the eCTD Requirements in SFDA?
Business Validation
This evaluation stage is a manual validation of the file after it passes electronic validation. It is a step before the actual evaluation. It aims to reduce the number of obvious file deficiencies usually discovered during the evaluation process.
The SFDA will validate (not evaluate) the presentation and main compliance issues, such as:
- Application type & fees
- MAH and Manufacturer legal status.
- Active Pharmaceutical Ingredient (API)
- Finished Pharmaceutical Product (FPP)
- Clinical Trials Data
- Safety data (SPC/PIL)
Approval Timelines
The drug sector has different assessment timelines depending on the application type and whether the product has acquired registration approval in a reference country. Read SFDA timelines.
Review and Approval Process
We find it essential to understand the SFDA approval process, namely the scientific assessment and the application journey through the drug sector departments. Knowing those details is necessary so we can put the registration requirements into context and help you determine which evaluation stage your application is at.
The drug approval process consists of multiple parallel evaluation routes within the drug sector. The parallel assessment allows departments to assess the application simultaneously. Below, we will go through each evaluation route in more detail.
Evaluation Department
Quality
- Active Pharmaceutical Ingredient (API)
- Finished Pharmaceutical Products (FPP)
Clinical Efficacy & Safety
- Clinical evaluation
- Bioequivalence (BE)
- Reference Safety Information (RSI)
- Summary of Product Characteristics (SPC)
- Patient Information Leaflet (PIL)
Inspection
The inspection department is responsible for evaluating, inspecting, and granting the SFDA GMP approval for drug manufacturers. Applicants should expect mandatory GMP licensing for all drug, health, herbal, and veterinary manufacturers. The licensing process includes a physical site inspection by SFDA inspectors and payment of the inspection fees.
Testing (SFDA LAB)
During the SFDA drug registration, you should expect a request for analysis samples and reference standards. Sometimes, you may be able to waive this request until the first commercial batch arrives in the Saudi market.
- Lab analysis
- Revision of related analysis documents
- Request drug samples and reference standards
- Testing commercial batches
Pricing
The drug pricing evaluation is the final stage of SFDA drug registration. It is mostly not parallel to the other assessments. The pricing department at the SFDA will require a price certificate and Economic Evaluation Studies (EES) to perform a pharmacoeconomic study of the drug and generate a report for the SFDA pricing committee. The committee will set the CIF price and ask the applicant to accept it or file a price appeal.
Registration Committee
The SFDA’s registration committee reviews all departments’ final comprehensive evaluation reports and then issues a formal approval or rejection decision.
Registration Certificate
Approved drug applications will receive a five-year registration certificate, entitling the company to market the product in Saudi Arabia.
Conclusion
After we understand that the drug application will undergo extensive assessment by SFDA experts, the applicant should expect multiple waves of inquiries (RFIs) throughout the process, particularly regarding Section 3.2.S, stability studies, clinical trials, bioequivalence, labeling (PIL/SPC), pharmacovigilance system, and QPPV.
Accelerated Review and Designations
There are many application pathways and designations that the SFDA offers to applicants. They are mostly available for new drug applications (NDA). Here are a few examples:
- Drug priority review procedure
- Verification and abridged registration
- Drug Special Access Program (SAP) in Saudi Arabia
- Orphan Drug Designation (ODD)
- Breakthrough Medicine Program
- Conditional Approval
Life Cycle Management (LCM)
Variation
The SFDA must review and approve the variation application before the applicant markets the product, whether it’s an administrative or technical change to the drug file. Read more: SFDA Variation Guidelines Overview.
Renewal
Companies must maintain the validity of their drug licenses at all times. When it is nearing expiry, they must submit a renewal application. Submission is allowed six months before the expiration date.
How PharmaKnowl Supports Registration Projects
We represent international MAH companies before the SFDA and provide direct regulatory services, including registration and product life-cycle management.
We create MAH accounts in SFDA, transfer their current approved applications from the distributors’ accounts, provide insights and guidance, and apply all available options to accelerate their access to the Saudi market. Contact us for an exploratory meeting to discuss your needs.
Read More:
About the Author

Regulatory affairs consultant, with more than 20 years of experience working for the SFDA, multinational companies, and as a consultant in PharmaKnowl.
Resources




Services
Events


