
About the Author: 穆罕默德·喬布蘭
在這篇文章中,我們概述了 SFDA 藥物註冊要求,並闡明瞭在沙烏地阿拉伯尋求藥物批准的審批程式。 我們將介紹不同的藥物申請類型及其要求,以説明您了解整個SFDA藥品註冊流程。
對於其他類型的產品,您可以參考:
介紹
沙特食品藥品管理局 (SFDA) 負責監管沙烏地阿拉伯的藥品,並確定藥品註冊要求和上市后義務。 自 2009 年 SFDA 採用 ICH 指南以來,這些要求已經發生了重大變化。 提交詳情已從紙質格式顯著過渡到 CTD,然後是 NeeS,最後是當前的 eCTD 檔案。 因此,為了説明您準備藥品檔案要求,瞭解漫長的註冊過程的細節也很重要。
提交計劃
準備提交 SFDA 的最佳實踐是制定監管策略。 此策略對於準確的業務決策和順利的註冊過程是必要的。 它提供了對即將對所有職能的監管影響的必要可見性。
由於合規性和註冊要求是監管策略的基礎,因此在開始註冊流程之前制定它最終將有助於您的專案取得積極進展,並使其執行更加有效和可靠。 閱讀: 如何為沙特市場制定監管策略。
藥品註冊要求
我收到的最常見的問題是,「SFDA 註冊要求是什麼? 這個問題很難用一個狹窄的清單來回答。 每個必需的檔都有其特定的合規措施,更不用說超出藥物檔案所需的全面盡職調查了。
因此,此處提供的資訊使監管專業人員能夠發現要求的深度和長度。 在這個方向上,我將這些相互關聯的過程分為四個部分:
- SFDA 指南
- 藥物檔案
- 藥物申請
- eCTD 提交
指引
根據藥物類型,您將在下面找到相關的 SFDA 藥品註冊指南:
人用藥物
- 人用藥物提交的數據要求
- 提交指南
- 人用藥品的有條件批准
- 優先審評指南
- SFDA 穩定性指南
- SFDA 藥物主檔 (DMF) 指南
- SFDA SPC、PIL 和標籤要求
- 醫藥產品的命名
- SFDA 防篡改包裝指南
- 藥品包裝平面設計
- 根據驗證和刪節註冊
- 模組 1 規範 生物等效性指南
- SFDA 生物仿製葯指南
- 研究性新藥 (IND) 要求
- 藥物參考標準
- SFDA 生物豁免指南
- 疫苗的生產和品質控制
草藥
- 草藥和健康產品提交的數據要求
- 草本和保健品的 PIL 和貼標資訊展示指南
- 含有維生素和礦物質的商品的一般規則
獸藥
- 獸藥商品的 SFDA VNeeS 規範
- 獸藥商品的數據要求
- 獸醫產品的 SFDA SPC、說明書和標籤
藥物檔案要求
檔案檔
- 新藥
這些是相當於 NDA 的創新藥,包括生物仿製葯。- 所有 5 個模組都是必需的。
- 仿製葯
- M1:所有部分
- M2:2.1、2.2、2.3、2.5.2
- M3:所有部分。
- M4: 不適用
- M5:僅限 5.1、5.2、5.3.1.2、5.3.1.3、5.3.1.4、5.3.7 和 5.4
- 保健品和草藥產品
- M1:所有部分。
- M3:所有部分。
- 獸藥:
- 第1部分
- 第2部分
- 第3部分
- 第 4 部分
達析報告格式
- 人類藥物:eCTD。
- 草藥和保健品:CTD 或 eCTD
- 獸藥:vNEES 或 CTD。
模組 1 要求
- 1.0 求職信
- 1.1 綜合目錄
- 1.2 申請表
- 1.3 產品資訊
- 1.3.1 產品特性 (SPC) 摘要
- 1.3.2 標註
- 1.3.3 患者資訊傳單 (PIL)
- 1.3.3.1 阿拉伯語傳單
- 1.3.3.2 英文宣傳單張
- 1.3.4 圖稿(模型)
- 1.3.5 採樣
- 1.4 專家資訊
- 1.4.1 品質
- 1.4.2 非臨床
- 1.4.3 臨床
- 1.5 環境風險評估
- 1.5.1 非轉基因生物 (Non-GMO)
- 1.5.2 轉基因生物
- 1.6 藥物警戒
- 1.6.1 藥物警戒系統
- 1.6.2 風險管理計劃
- 1.7 證書和檔
- 1.7.1 GMP 證書
- 1.7.2 CPP 或免費銷售
- 1.7.3 分析證書 – 原料葯/成品
- 1.7.4 分析證書 – 輔料
- 1.7.5 酒精含量聲明
- 1.7.6 豬肉含量聲明
- 1.7.7 TSE 適宜性證書
- 1.7.8 產品配方中的稀釋劑和著色劑
- 1.7.9 專利資訊
- 1.7.10 DMF 的訪問或確認信
- 1.8 定價
- 1.8.1 價格表
- 1.8.2 其他相關文檔
- 1.9 對問題的回答
藥物申請
申請人必須填寫藥品註冊申請並通過沙特藥品註冊 (eSDR) 系統提交。 它是一個可供沙特當地公司使用的入口網站;它使申請人能夠執行以下操作:
- 填寫並匯出模組 1 的應用程式。
- 支付申請費。
- 提交檔案。
- 接收 SFDA 查詢 (RFI)。
- 接收 SFDA 決策。
- 列印註冊證書。
- 提交變更和續訂(生命週期管理)。
應用程式類型
eSDR 中的藥物應用分為三大類型,亞型更多,如下:
- 人用藥品
- 新藥
- 生物
- 放射性藥物
- 仿製葯(多源)藥物
- 健康 / 草藥產品
- 獸用產品
- 新藥
- 生物葯
- 仿製葯(多源)藥物
- 草藥產品
- 健康產品
申請費用
有一個與藥品註冊相關的費用矩陣,我們在我們的帖子中詳細介紹了: SFDA 費用.
提交
下圖顯示了應用程式提交過程,其中涉及兩層驗證。
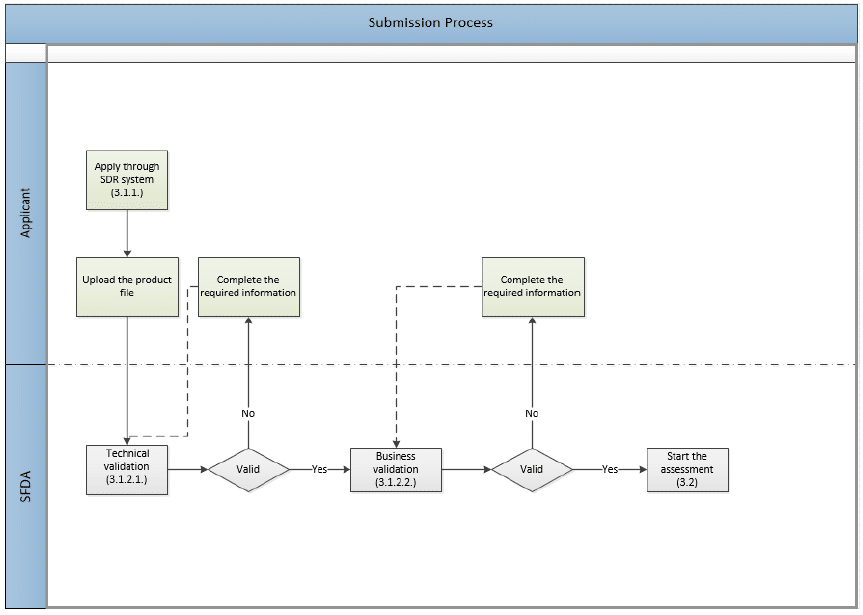
驗證
技術驗證
SFDA 收到提交的檔案,並根據 SFDA eCTD 驗證標準對編譯的 eCTD 進行電子驗證。
業務驗證
此評估階段是在通過電子驗證后對文件執行的手動驗證。 這是開始實際評估之前的一個步驟。 它旨在減少通常在評估過程中發現的明顯檔缺陷的數量。
SFDA 將驗證(而不是評估)演示文稿和主要合規性問題,例如:
- 申請類型和費用
- MAH 和製造商的法律地位。
- 活性藥物成分 (API)
- 成品藥 (FPP)
- 臨床試驗 數據
- 安全資料 (SPC/PIL)
審批時程表
藥品行業的評估時程表不同,具體取決於申請類型以及商品是否已在 參考國家/地區獲得註冊批准。閱讀 SFDA 時程表。
審核和批准流程
我們發現瞭解 SFDA 審批流程至關重要,即科學評估和通過藥品行業部門的申請過程。 瞭解這些詳細資訊是必要的,這樣我們才能將註冊要求放在上下文中,並説明您確定您的申請處於哪個評估階段。
藥物審批流程包括藥品行業內的多個平行評估途徑。 並行評估允許部門同時評估應用程式。 下面,我們將更詳細地介紹每條評估路線。
評估部門
品質
- 活性藥物成分 (API)
- 成品藥 (FPP)
臨床療效和安全性
- 臨床評估
- 生物等效性 (BE)
- 參考安全資訊 (RSI)
- 產品特性 (SPC) 摘要
- 患者資訊傳單 (PIL)
檢查
檢查部門負責對藥品生產企業進行評估、檢查和授予 SFDA GMP批准 。申請人應期望所有藥品、保健品、草藥和獸醫製造商都獲得強制性 GMP 許可。許可流程包括 SFDA 檢查員的物理現場檢查和支付 檢查費用。
測試 (SFDA LAB)
在 SFDA 藥品註冊期間,您應該會收到分析樣品和參考標準品的要求。 有時,在第一批商業產品運抵沙特市場之前,您可以放棄此請求。
- 實驗室分析
- 修訂相關分析檔
- 索取藥物樣品和參考標準品
- 測試商業批次
定價
定價評估是 SFDA 藥品註冊的最後一個階段。 它與其他評估不同。 該部門對藥物進行藥物經濟學研究,併為 SFDA 定價委員會生成報告。 委員會將設定 CIF 價格,並要求申請人接受或上訴。
註冊委員會
SFDA 註冊委員會審查所有部門的最終綜合評價報告,然後出具正式的批准或拒絕決定。
註冊證書
獲得批准的藥物申請將獲得註冊證書,使公司有權在沙烏地阿拉伯進行行銷。 此證書的有效期為 5 年。
結論
在我們瞭解到藥物申請將經過 SFDA 專家的廣泛評估后,申請人應該會在此過程中收到多波詢問 (RFI),尤其是關於 API、 穩定性、臨床試驗、生物等效性、 標籤 (PIL/SPC)、藥物警戒系統和 QPPV。
加速審批途徑
其他航線和名稱
- 孤兒葯資格認定 (ODD)
- 有條件批准
- 突破性藥物認定
生命週期管理 (LCM)
變化
無論是對藥品檔案的行政還是技術變更,SFDA 都必須在申請人銷售產品之前審查和批准變更申請。 閱讀更多: SFDA 變體指南概述。
更新
公司必須始終保持藥品許可證的有效性。 當合同即將到期時,他們必須提交續期申請。 允許在到期日期前 6 個月提交。



