
Acerca del autor

Editor de reglamentación
Publicado por el equipo de asuntos regulatorios de PharmaKnowl, oficina de Riad.
La SFDA tiene estrictos requisitos de etiquetado y empaquetado que las empresas deben cumplir durante el proceso de registro de SFDA . Una vez aprobado, la información del etiquetado no debe modificarse mínimamente para garantizar la conformidad del producto y evitar el rechazo o la retirada del mercado.
Este artículo revisará los requisitos de etiquetado para medicamentos y dispositivos médicos.
Tabla de contenido
Etiquetado de medicamentos
Los requisitos específicos de etiquetado SFDA deben aplicarse a todos los componentes del diseño del medicamento, incluyendo el envase, la etiqueta, el papel de aluminio, el prospecto (Folleto de Información para el Paciente) y el Resumen de las Características del Producto (RCP). A continuación, se describen los requisitos generales para cada uno de los componentes mencionados, ya que constituyen uno de los principales requisitos de registro de medicamentos en Arabia Saudita .
Embalaje exterior e interior (embalaje secundario)
Nombre del medicamento
La descripción del producto debe aparecer en más de tres caras no opuestas de las seis caras de la caja. Debe estar escrita en inglés y árabe.
Declaración de la(s) sustancia(s) activa(s)
La expresión del principio activo debe presentarse cualitativa y cuantitativamente por unidad de dosificación. Debe estar escrita en inglés y árabe.
Declaración de indicación
La indicación del producto debe estar resaltada en el envase, por ejemplo, para el tratamiento de, la prevención de, etc.
Lista de excipientes
Los excipientes con efectos o acciones reconocidos deben expresarse cualitativamente. Todos los excipientes deben indicarse si el medicamento es parenteral, tópico, oftálmico o inhalatorio.
Forma farmacéutica y contenido
El contenido debe mencionarse por peso, volumen, número de dosis o número de unidades de administración (por ejemplo, 10 comprimidos, 100 ml, etc.).
Método y vía(s) de administración
El método de administración y las instrucciones para el uso correcto del medicamento deben estar claramente indicados, por ejemplo, «Agitar bien antes de usar». Para las instrucciones de uso, se puede consultar el prospecto adjunto.
Advertencia especial
Se debe resaltar una advertencia especial en la etiqueta, por ejemplo, «Proteger de la luz solar, Mantener fuera de la vista y del alcance de los niños, sólo para uso externo».
Condición de almacenamiento
Se deben mencionar las condiciones de almacenamiento del producto, por ejemplo, almacenar por debajo de 25 ° C o almacenar por debajo de 30 ° C. Se debe agregar la estabilidad en uso para productos multidosis, por ejemplo, la vida útil después de la primera apertura debe ser de un mes, dos meses, tres meses, etc. Debe reflejar lo que se indica en el estudio de estabilidad SFDA .
Fabricante y MAH
Se debe mencionar el nombre y la dirección del fabricante y del titular de la autorización de comercialización.
Blisters o tiras (Embalaje Primario)
Nombre del medicamento
El nombre y la concentración del producto deben aparecer en cada blíster; si estos son demasiado pequeños, la información debe repetirse en un patrón a lo largo de toda la tira. Se requiere en inglés y árabe, requisitos obligatorios de etiquetado SFDA para todas las piezas.
Nombre del titular de la autorización de comercialización
La marca solamente es suficiente.
Fechas de fabricación y caducidad
Las fechas deben expresarse, por ejemplo, 02/2010 o febrero 2010.
Número de lote
El número de lote y la fecha de caducidad deben estar al final de cada tira blíster.
Resumen de las características del producto (SPC)
Aunque el RCP o la Ficha Técnica no se imprimen ni se adjuntan al medicamento, deben aplicarse los requisitos de etiquetado de SFDA . Solo la Ficha Técnica aprobada puede distribuirse a los profesionales sanitarios y utilizarse en materiales promocionales.
Información primaria
Nombre del medicamento, dosis y forma farmacéutica.
Composición cualitativa y cuantitativa
El principio activo debe expresarse como dosis por unidad. Se debe incluir una declaración estándar para los excipientes, por ejemplo: «Para obtener una lista completa de excipientes, consulte la sección «Lista de excipientes»».
Forma farmacéutica
Se debe mencionar la forma farmacéutica del producto (p. ej., comprimido recubierto con película, comprimido de liberación prolongada, etc.), junto con una descripción de la misma. Si los comprimidos tienen una ranura, se debe indicar si esta facilita su fragmentación o si permite dividirlos en dosis iguales.
Datos clínicos
Indicaciones terapéuticas
La indicación debe definir la enfermedad o condición objetivo para el tratamiento o prevención y su indicación en adultos, neonatos, lactantes, niños y adolescentes de edad (meses, años).
Posología y forma de administración
Las recomendaciones de dosis deben especificarse por intervalo de dosis para cada categoría, cuando corresponda (determinar la edad, el peso y la superficie corporal de los subgrupos de la población, según corresponda). Se debe proporcionar información sobre poblaciones especiales, como pacientes de edad avanzada, insuficiencia renal, insuficiencia hepática y población pediátrica. Asimismo, se deben proporcionar medidas preventivas para la manipulación y administración del producto.
Contraindicaciones
Son casos en los que no se deben administrar medicamentos por razones de seguridad, es decir, hipersensibilidad al principio activo o a alguno de los excipientes.
Advertencias y precauciones especiales de empleo
Información sobre riesgos específicos, medidas específicas de minimización de riesgos, reacciones adversas, información de seguridad,
Interacciones
Estudio de interacción clínica (in vivo), Interacción farmacodinámica, Interacción farmacocinética.
Fertilidad, embarazo y lactancia
Las recomendaciones de uso en mujeres embarazadas y lactantes y mujeres en edad fértil deben basarse en ensayos clínicos , estudios no clínicos y actividad farmacológica.
Efectos sobre la capacidad para conducir y utilizar máquinas
Efectos indeseables
Resumen del perfil de seguridad, reacciones adversas y dirección de la autoridad nacional para notificar cualquier efecto secundario.
Sobredosis
Efecto de diferentes niveles de dosificación tomados accidentalmente, por error o por intentos de suicidio.
Propiedades farmacológicas
El contenido debe mencionarse por peso, volumen, número de dosis o número de unidades de administración del medicamento (por ejemplo, 10 comprimidos, 100 ml, etc.).
Datos farmacéuticos
- Lista de excipientes
- Incompatibilidades
- Duración
- Precauciones especiales de conservación
- Naturaleza y contenido del envase.
Precauciones especiales de eliminación y manipulación
Requisitos especiales para la eliminación; si no se especifica «no se requieren requisitos especiales», se puede indicar. La eliminación debe realizarse conforme a las normativas locales.
Titular de la autorización de comercialización
Número de autorización de comercialización
Fecha de la primera Autorización/Renovación de la Autorización
Fecha de revisión del texto
Folleto de información para el paciente (PIL)
¿Qué es (nombre inventado) y para qué se utiliza?
Nombre de invención, concentración, forma farmacéutica, ingrediente activo, indicación terapéutica y beneficio del uso de este medicamento.
Antes de tomar o utilizar el producto
Contraindicaciones, precauciones adecuadas de uso/advertencias especiales, interacciones con otros medicamentos/alimentos/bebidas, información para uso en mujeres embarazadas o en periodo de lactancia, información sobre fertilidad, conducción y uso de máquinas y advertencias sobre excipientes.
Cómo tomar o utilizar el producto
Posología, Modo y vía(s) de administración, Frecuencia de administración, Instrucciones de uso adecuado, Duración del tratamiento, Sobredosis, Uso irregular del medicamento,
Posibles efectos secundarios
Descripción de efectos secundarios, efectos secundarios adicionales en niños y adolescentes.
Cómo almacenar el nombre del producto
Se requieren direcciones y precauciones especiales durante el almacenamiento y las condiciones de conservación.
Más información
Información sobre el principio(s) activo(s) y el excipiente(s), forma farmacéutica, naturaleza y contenido del envase, nombre y dirección del titular de la autorización de comercialización y del fabricante responsable de la liberación del lote, fecha de revisión del prospecto y dirección de la autoridad nacional para notificar cualquier efecto adverso.
Información del titular de la autorización de comercialización
Nombre y dirección
Información del fabricante
Nombre y dirección
Este folleto fue revisado por última vez el
Fecha: Mes y Año
Para informar sobre cualquier efecto secundario
Añadir en esta sección toda la información de farmacovigilancia y QPPV e instrucciones sobre cómo informar.
Etiquetado de dispositivos médicos
Requisitos de etiquetado SFDA para dispositivos médicos Se aplican a los materiales de embalaje, las etiquetas y las instrucciones de uso (IFU) y deben presentarse en consecuencia en las solicitudes de registro de dispositivos médicos (MDMA). Se aplican tanto a los de bajo riesgo y dispositivos médicos de alto riesgo.
El término etiquetado es un término colectivo que comprende:
- La etiqueta,
- Instrucciones de uso (a veces denominadas manual del operador) y
- Cualquier otra información que esté relacionada con la identificación, descripción técnica, finalidad prevista y uso adecuado del dispositivo médico,
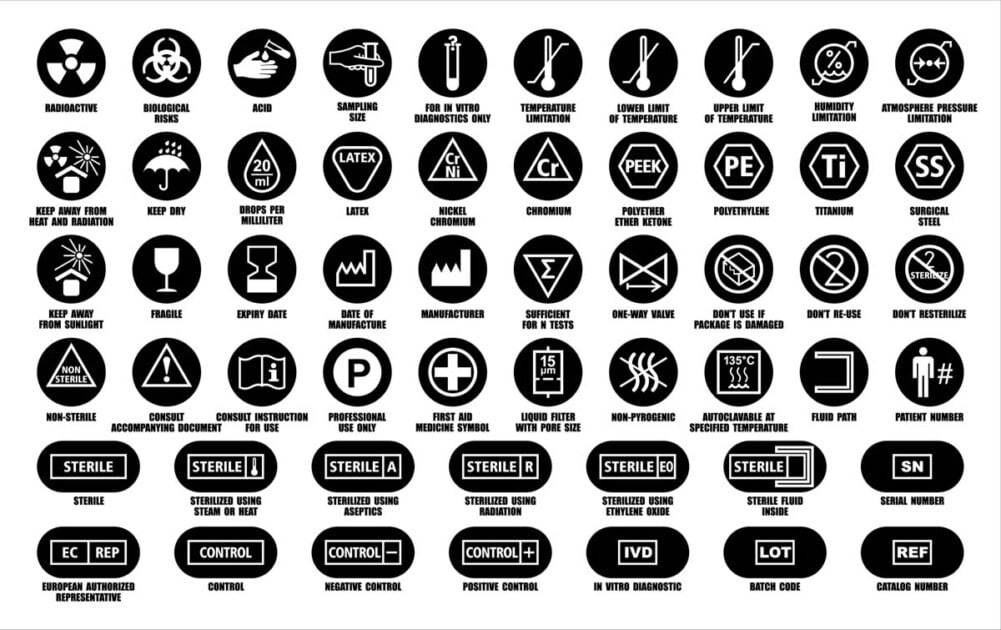
El término etiqueta describe información escrita, impresa o gráfica que es:
- Fijado o que aparece en el propio dispositivo médico (incluida la pantalla electrónica)
- En el embalaje de:
- Cada unidad (envoltorio)
- Múltiples dispositivos (contenedores)
- En un prospecto (se utiliza cuando no es práctico o apropiado colocar una etiqueta directamente en el dispositivo médico. No es práctico cuando las limitaciones físicas impiden que esto suceda).
El objetivo principal del etiquetado es:
- Identificar el dispositivo médico y su fabricante,
- Describa el uso y el rendimiento previstos del dispositivo.
- Describir cómo se debe utilizar, mantener y almacenar el dispositivo, y
- Proporcionar información sobre cualquier riesgo residual del dispositivo, advertencias, limitaciones o contraindicaciones.
Instrucciones de uso (IFU)
El término «Instrucciones de uso (IFU)» significa la información proporcionada por el fabricante para informar al usuario del dispositivo:
- Del propósito previsto y uso adecuado del dispositivo médico.
- De las precauciones que se deben tomar.
Es posible que no se necesiten instrucciones de uso o que se abrevien para los dispositivos si se pueden usar de manera segura y según lo previsto por el fabricante sin dichas instrucciones de uso (por ejemplo, lentes ópticas, bastones para caminar o simples apósitos para heridas).
Lenguaje de etiquetado
Cuando el usuario del dispositivo médico sea:
- Es probable que esté calificado profesionalmente y el etiquetado deberá estar en inglés.
- Para el público general, la etiqueta y las instrucciones de uso deberán estar en árabe e inglés siempre que sea posible. De no ser posible, el idioma utilizado en la etiqueta y las instrucciones de uso será el árabe.
En ambas situaciones, el texto deberá redactarse en términos fácilmente comprensibles para el usuario previsto, acordes con sus conocimientos técnicos, experiencia, educación o capacitación.
Idioma de las instrucciones del fabricante
Las instrucciones para la manipulación, el almacenamiento, el transporte, la instalación, el mantenimiento y la eliminación de los dispositivos médicos deberán estar en inglés y, cuando corresponda, en árabe. El texto deberá estar redactado en términos fácilmente comprensibles para el usuario previsto, acorde con sus conocimientos técnicos, experiencia, formación o capacitación, en caso de que personas sin cualificación médica puedan realizar dicha tarea. Si el dispositivo está diseñado para personas no cualificadas, las instrucciones para la manipulación, el almacenamiento, el transporte y el mantenimiento de los dispositivos médicos deberán estar en árabe e inglés.
Medio de etiquetado
Las etiquetas se proporcionarán en un formato legible para humanos, pero podrán complementarse con formatos legibles por máquina, como identificación por radiofrecuencia (RFID) o códigos de barras.
Contenido del etiquetado
Nombre del dispositivo
La marca o el nombre comercial del producto deben estar impresos en la etiqueta. Asimismo, el número de modelo del producto está impreso en la etiqueta.
Nombre y dirección del fabricante
El nombre y la dirección del fabricante deben estar impresos en las etiquetas y deben coincidir letra por letra con la dirección mencionada en el acuerdo de autorización de comercialización presentado y la posterior licencia de autorización de comercialización. Cualquier discrepancia en la dirección resultaría en la devolución de la solicitud presentada a SFDA , lo que dificultaría la obtención de la autorización de comercialización del dispositivo médico.
Fabricante legal en casos OEM/OBL
OEM es la abreviatura de fabricante de equipo original, mientras que OBL es etiquetado de marca propia.
En casos de fabricantes OEM/OBL, donde se ha seleccionado la jurisdicción de la UE como base para la solicitud de MDMA, SFDA considera como fabricante legal el nombre del fabricante junto al «Símbolo del Fabricante» en la etiqueta. Si esto no aplica, se deben presentar los siguientes documentos a SFDA :
- Copia de los certificados de evaluación de la conformidad basados en la ruta de evaluación de la conformidad utilizada (es decir, certificado CE, certificado DE, certificado de garantía de calidad de producción, etc.)
- Copia de la «Declaración de conformidad del producto».
- Copia del etiquetado (incluidas las instrucciones de uso)
- Informe de auditoría más reciente (si corresponde)
Fuente de alimentación
Cuando el dispositivo esté conectado a una fuente de alimentación de CA, una indicación de la frecuencia nominal (60 Hertz) y los valores de voltaje con sus tolerancias para los que ha sido diseñado el producto.
Etiquetado de diagnóstico in vitro
Esto indica que el producto es para uso diagnóstico in vitro si es un dispositivo médico IVD.
Condiciones de almacenamiento
Cuando corresponda, una indicación de cualquier condición especial de almacenamiento y manipulación que se aplique.
Advertencias o precauciones
Cualquier advertencia, precaución, limitación o contraindicación.
Número de lote o lote
El código de lote/número de LOTE o el número de serie permiten aplicar medidas de cumplimiento posteriores a la comercialización si es necesario rastrear o retirar el producto. Sin embargo, en el caso de los accesorios de los dispositivos médicos de diagnóstico in vitro, este código puede sustituirse por un número de control y, en el caso del software, debe sustituirse por un número de versión.
Fecha de caducidad
Una indicación inequívoca de la fecha hasta la cual el producto puede usarse de forma segura (por ejemplo, en productos desechables estériles o de un solo uso) cuando sea pertinente.
Cantidad
Cuando sea pertinente, se expresará una indicación de la cantidad neta del contenido en términos de peso o volumen, recuento numérico o cualquier combinación de estos u otros términos que reflejen con precisión el contenido del paquete.
De uso único o múltiple
Si el producto está destinado a un solo uso, una indicación de tal hecho.



