
Über den Autor

Regulierungsredakteur
Veröffentlicht vom Regulatory Affairs Team von PharmaKnowl, Büro in Riad.
Die SFDA hat strenge Kennzeichnungs- und Verpackungsvorschriften, die Unternehmen im Rahmen des SFDA einhalten müssen. Nach der Zulassung dürfen die Kennzeichnungsinformationen nicht einmal geringfügig verändert werden, um die Produktkonformität zu gewährleisten und eine Ablehnung der Lieferung oder Rücknahme vom Markt zu vermeiden.
In diesem Artikel werden die Kennzeichnungsanforderungen für Arzneimittel und Medizinprodukte untersucht.
Inhaltsverzeichnis
Arzneimittelkennzeichnung
Die spezifischen Kennzeichnungsanforderungen SFDA gelten für alle Bestandteile der Arzneimittelgrafik, einschließlich Karton, Etikett, Folie, Beipackzettel (Patienteninformationsblatt) und Zusammenfassung der Merkmale des Arzneimittels (SPC). Wir erläutern hier die wichtigsten Anforderungen für jeden der oben genannten Bestandteile, da sie zu den wichtigsten Anforderungen für die Arzneimittelzulassung in Saudi-Arabien gehören.
Außen- und Innenverpackung (Sekundärverpackung)
Name der Medizin
Die Produktbeschreibung muss auf mehr als drei der sechs Seiten der Schachtel, die sich nicht gegenüberliegen, erscheinen. Sie sollte in Englisch und Arabisch verfasst sein.
Angabe des/der Wirkstoffs/Wirkstoffe
Die Wirkstoffausprägung sollte qualitativ und quantitativ pro Dosierungseinheit dargestellt werden. Die Formulierung sollte in englischer und arabischer Sprache erfolgen.
Indikationserklärung
Die Produktindikation sollte auf der Packung hervorgehoben sein. z. B. zur Behandlung von, zur Vorbeugung von usw.
Liste der sonstigen Bestandteile
Hilfsstoffe mit anerkannten Wirkungen sollten qualitativ angegeben werden. Alle Hilfsstoffe müssen angegeben werden, wenn das Arzneimittel parenteral, topisch, zur Augenbehandlung oder zur Inhalation verwendet wird.
Darreichungsform und Inhalt
Der Inhalt sollte nach Gewicht, Volumen, Anzahl der Dosen oder Anzahl der Verabreichungseinheiten (z. B. 10 Tabletten, 100 ml usw.) angegeben werden.
Art und Weg der Verabreichung
Die Art der Verabreichung und die Anweisungen zur ordnungsgemäßen Anwendung des Arzneimittels sollten klar angegeben werden, z. B. „Vor Gebrauch gut schütteln“. Die Gebrauchsanweisung finden Sie in der beiliegenden Packungsbeilage.
Besondere Warnung
Auf dem Etikett sollte ein besonderer Warnhinweis hervorgehoben werden, z. B. „Vor Sonnenlicht schützen. Außerhalb der Sicht- und Reichweite von Kindern aufbewahren. Nur zur äußerlichen Anwendung.“
Lagerbedingungen
Die Lagerbedingungen des Produkts sollten erwähnt werden, z. B. „Unter 25 ° C lagern“ oder „Unter 30 ° C lagern“. Bei Mehrdosenprodukten sollte die Stabilität während des Gebrauchs hinzugefügt werden, z. B. sollte die Haltbarkeitsdauer nach dem ersten Öffnen einen Monat, zwei Monate, drei Monate usw. betragen. Sie muss den Angaben in der Stabilitätsstudie SFDA entsprechen.
Hersteller und Zulassungsinhaber
Name und Adresse des Herstellers und des Inhabers der Genehmigung für das Inverkehrbringen sollten angegeben werden.
Blister oder Streifen (Primärverpackung)
Name der Medizin
Name und Stärke des Produkts sollten über jeder Blistertasche angegeben sein. Sind die Taschen zu klein, sollten die Informationen in einem Muster über den gesamten Streifen wiederholt werden. Die Angaben sind in Englisch und Arabisch erforderlich, da dies für SFDA -Kennzeichnung aller Teile verbindlich ist.
Name des Zulassungsinhabers
Der bloße Markenname ist ausreichend.
Herstellungs- und Verfallsdaten
Daten sollten beispielsweise als „02/2010“ oder „Februar 2010“ angegeben werden.
Chargennummer
Die Chargennummer (Losnummer) und das Verfallsdatum sollten am Ende jedes Blisterstreifens stehen.
Zusammenfassung der Merkmale des Arzneimittels (SPC)
Obwohl die SPC oder SmPC nicht gedruckt und dem Arzneimittel beigefügt ist, müssen die Kennzeichnungsvorschriften SFDA eingehalten werden. Nur die genehmigte SmPC darf an die Ärzte verteilt und für Werbematerialien verwendet werden.
Primärinformationen
Name des Arzneimittels, Stärke und Darreichungsform.
Qualitative und quantitative Zusammensetzung
Der Wirkstoff sollte als Dosis pro Einheit angegeben werden. Für Hilfsstoffe sollte eine Standardangabe enthalten sein, z. B.: „Eine vollständige Liste der Hilfsstoffe finden Sie im Abschnitt ‚Liste der Hilfsstoffe‘.“
Darreichungsform
Die Darreichungsform des Produkts sollte angegeben werden, z. B. Filmtablette, Retardtablette usw., zusammen mit einer Beschreibung der Darreichungsform. Wenn die Tabletten mit einer Bruchkerbe versehen sind, sollte angegeben werden, ob diese das Teilen erleichtert oder die Tabletten in gleiche Dosen aufteilt.
KLINISCHE ANGABEN
Therapeutische Hinweise
Die Indikation sollte die Zielkrankheit oder den Zielzustand für die Behandlung oder Prävention und ihre Indikation bei Erwachsenen, Neugeborenen, Säuglingen, Kindern und Jugendlichen im Alter (Monate, Jahre) definieren.
Dosierung und Art der Anwendung
Dosierungsempfehlungen sollten gegebenenfalls für jede Kategorie pro Dosierungsintervall angegeben werden (Alter, Gewicht und Körperoberfläche von Teilmengen der Bevölkerung sind gegebenenfalls zu bestimmen). Informationen zu besonderen Patientengruppen wie älteren Patienten, Patienten mit Nieren- oder Leberfunktionsstörungen sowie Kindern und Jugendlichen sollten bereitgestellt werden. Außerdem sollten vorbeugende Maßnahmen für die Handhabung und Verabreichung des Produkts angegeben werden.
Kontraindikationen
Dabei handelt es sich um Fälle, in denen Arzneimittel aus Sicherheitsgründen nicht verabreicht werden dürfen, beispielsweise bei einer Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile.
Besondere Warnhinweise und Vorsichtsmaßnahmen
Informationen über spezifische Risiken, spezifische Maßnahmen zur Risikominimierung, Nebenwirkungen, Sicherheitsinformationen,
Interaktionen
Klinische Interaktionsstudie (in vivo), Pharmakodynamische Interaktion, Pharmakokinetische Interaktion.
Fruchtbarkeit, Schwangerschaft und Stillzeit
Empfehlungen zur Anwendung bei schwangeren und stillenden Frauen sowie Frauen im gebärfähigen Alter sollten auf klinischen Studien , nichtklinischen Studien und der pharmakologischen Aktivität basieren.
Auswirkungen auf die Fähigkeit, Maschinen zu fahren und zu benutzen
Nebenwirkungen
Zusammenfassung des Sicherheitsprofils, der Nebenwirkungen und Adresse der nationalen Behörde zur Meldung etwaiger Nebenwirkungen.
Überdosis
Auswirkungen unterschiedlicher Dosierungen bei versehentlicher, irrtümlicher oder Selbstmordversuchseinnahme.
Pharmakologische Eigenschaften
Der Inhalt sollte nach Gewicht, Volumen, Anzahl der Dosen oder Anzahl der Verabreichungseinheiten des Arzneimittels (z. B. 10 Tabletten, 100 ml usw.) angegeben werden.
Pharmazeutische Angaben
- Liste der sonstigen Bestandteile
- Interaktion
- Haltbarkeit
- Besondere Vorsichtsmaßnahmen für die Aufbewahrung
- Art und Inhalt des Behälters.
Besondere Vorsichtsmaßnahmen für die Entsorgung und Handhabung
Besondere Anforderungen für die Entsorgung, sofern keine besonderen Anforderungen genannt werden können. Die Entsorgung sollte gemäß den örtlichen Vorschriften erfolgen.
Inhaber der Genehmigung für das Inverkehrbringen
Zulassungsnummer
Datum der Erstzulassung/Erneuerung der Zulassung
Datum der Überarbeitung des Textes
Patienteninformationsbroschüre (PIL)
Was ist (Phantasiename) und wofür wird es angewendet?
Der erfundene Name, die Stärke, die Darreichungsform, der Wirkstoff, die therapeutische Indikation und der Nutzen der Anwendung dieses Arzneimittels.
Bevor Sie das Produkt einnehmen oder verwenden
Gegenanzeigen, Vorsichtsmaßnahmen für die Anwendung/besondere Warnhinweise, Wechselwirkungen mit anderen Arzneimitteln/Nahrungsergänzungsmitteln/Getränken, Informationen zur Anwendung bei schwangeren oder stillenden Frauen, Informationen zur Fruchtbarkeit, zum Führen von Fahrzeugen und zum Bedienen von Maschinen sowie Warnhinweise zu sonstigen Bestandteilen.
Wie ist das Produkt einzunehmen oder anzuwenden?
Dosierung, Art und Weg der Verabreichung, Häufigkeit der Verabreichung, Hinweise zur richtigen Anwendung, Dauer der Behandlung, Überdosierung, unregelmäßige Einnahme des Arzneimittels,
Mögliche Nebenwirkungen
Beschreibung der Nebenwirkungen, zusätzliche Nebenwirkungen bei Kindern und Jugendlichen.
So speichern Sie den Produktnamen
Bei der Lagerung und den Lagerungsbedingungen sind besondere Hinweise und Vorsichtsmaßnahmen erforderlich.
Weitere Hinweise
Informationen zu Wirkstoff(en) und Hilfsstoff(en), Darreichungsform, Art und Inhalt des Behältnisses, Name und Anschrift des Inhabers der Genehmigung für das Inverkehrbringen und des für die Chargenfreigabe verantwortlichen Herstellers, Überarbeitungsdatum der Packungsbeilage und Anschrift der nationalen Behörde zur Meldung etwaiger Nebenwirkungen.
Informationen für Inhaber der Zulassung
Name und Adresse
Herstellerinformationen
Name und Adresse
Diese Packungsbeilage wurde zuletzt überarbeitet am
Datum: Monat und Jahr
Um Nebenwirkungen zu melden
Fügen Sie in diesem Abschnitt alle Informationen zur Pharmakovigilanz und QPPV sowie Anweisungen zur Meldung hinzu.
Kennzeichnung medizinischer Geräte
Die SFDA Kennzeichnungsanforderungen für Medizinprodukte gelten für Verpackungsmaterialien, Etiketten und Gebrauchsanweisungen (IFU) und müssen entsprechend in den Zulassungsanträgen für Medizinprodukte (MDMA) eingereicht werden. Sie gelten sowohl für risikoarme und medizinische Geräte mit hohem Risiko.
Der Begriff Kennzeichnung ist ein Sammelbegriff und umfasst:
- Das Etikett,
- Gebrauchsanweisung (manchmal auch als Benutzerhandbuch bezeichnet) und
- Alle anderen Informationen, die sich auf die Identifizierung, die technische Beschreibung, den Verwendungszweck und die ordnungsgemäße Verwendung des Medizinprodukts beziehen,
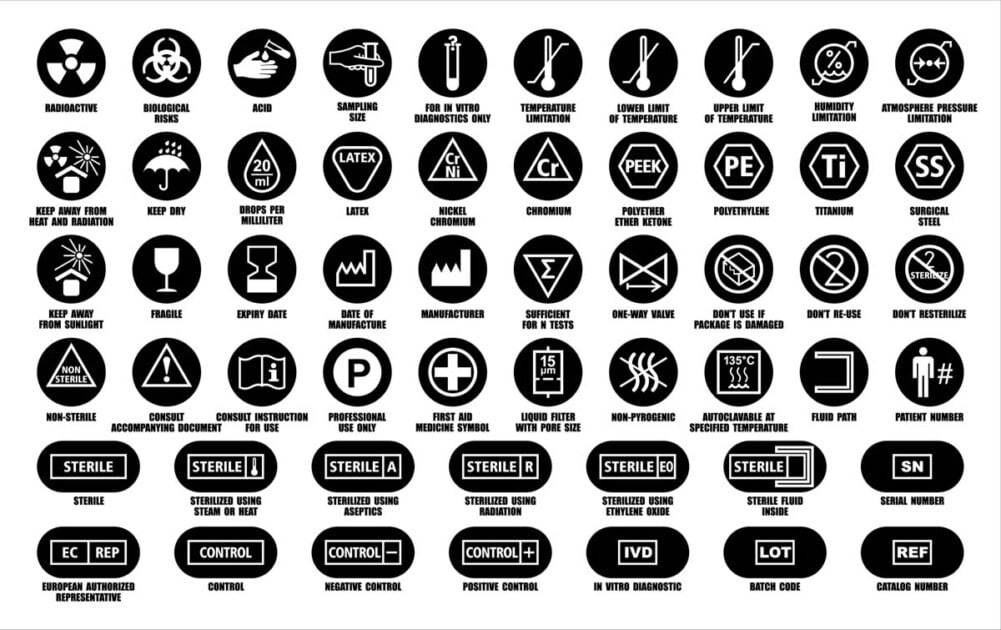
Der Begriff Etikett beschreibt schriftliche, gedruckte oder grafische Informationen, die:
- Am Medizinprodukt selbst angebracht oder auf diesem sichtbar (einschließlich elektronischer Anzeige)
- Auf der Verpackung von:
- Jede Einheit (Verpackung)
- Mehrere Geräte (Container)
- Auf einem Beipackzettel (wird verwendet, wenn es unpraktisch oder unangemessen ist, ein Etikett direkt auf dem Medizinprodukt anzubringen. Unpraktisch bedeutet, wenn physikalische Einschränkungen dies verhindern).
Der Hauptzweck der Kennzeichnung besteht darin:
- Identifizierung des Medizinprodukts und seines Herstellers,
- Beschreiben Sie den Verwendungszweck und die Leistung des Geräts.
- Beschreiben Sie, wie das Gerät verwendet, gewartet und gelagert werden soll, und
- Geben Sie Informationen zu etwaigen verbleibenden Risiken, Warnungen, Einschränkungen oder Kontraindikationen des Geräts an.
Gebrauchsanweisung (IFU)
Der Begriff „Gebrauchsanweisung (IFU)“ bezeichnet die vom Hersteller bereitgestellten Informationen, die den Geräteanwender informieren:
- Über die Zweckbestimmung und den bestimmungsgemäßen Gebrauch des Medizinprodukts.
- Von allen zu treffenden Vorsichtsmaßnahmen.
Für Geräte ist eine IFU möglicherweise nicht erforderlich oder muss abgekürzt werden, wenn sie ohne eine solche Gebrauchsanweisung sicher und wie vom Hersteller vorgesehen verwendet werden können (z. B. optische Linsen, Gehstöcke oder einfache Wundverbände).
Beschriftungssprache
Wenn der Benutzer des Medizinprodukts:
- Vorausgesetzt, die Beschriftung ist fachlich qualifiziert, erfolgt sie in englischer Sprache.
- Für Laien: Das Etikett und die Gebrauchsanweisung sollten, soweit möglich, auf Arabisch und Englisch verfasst sein. Wo dies nicht möglich ist, sollte Arabisch als Sprache für das Etikett und die Gebrauchsanweisung verwendet werden.
In beiden Fällen muss der Text in einer für den vorgesehenen Benutzer leicht verständlichen Sprache verfasst sein, die seinem technischen Wissen, seiner Erfahrung, Ausbildung oder Schulung entspricht.
Sprache der Herstelleranweisungen
Anweisungen für Handhabung, Lagerung, Transport, Installation, Wartung und Entsorgung der Medizinprodukte müssen in englischer Sprache und, soweit gerechtfertigt, in arabischer Sprache verfasst sein. Der Text muss in einer für den vorgesehenen Anwender leicht verständlichen Sprache verfasst sein und seinen technischen Kenntnissen, Erfahrungen, Ausbildungen oder Schulungen entsprechen, sofern Personen ohne medizinische Qualifikation solche Arbeiten durchführen dürfen. Ist das Produkt für Laien konzipiert, müssen die Anweisungen für Handhabung, Lagerung, Transport und Wartung der Medizinprodukte in arabischer und englischer Sprache verfasst sein.
Kennzeichnungsmedium
Etiketten müssen in einem für Menschen lesbaren Format bereitgestellt werden, können jedoch durch maschinenlesbare Formen wie Radiofrequenz-Identifikation (RFID) oder Strichcodes ergänzt werden.
Inhalt der Kennzeichnung
Gerätename
Der Handels- oder Markenname des Produkts sollte auf dem Etikett aufgedruckt sein. Ebenso ist die Modellnummer des Produkts auf dem Etikett aufgedruckt.
Name und Anschrift des Herstellers
Name und Anschrift des Herstellers müssen auf den Etiketten angegeben sein und müssen buchstabengetreu mit den Adressangaben in der eingereichten AR-Vereinbarung und der nachfolgenden AR-Lizenz übereinstimmen. Abweichungen in den Adressangaben führen zur Rücksendung des eingereichten Antrags an SFDA und können somit die Zulassung des Medizinprodukts erschweren.
Rechtmäßiger Hersteller in OEM/OBL-Fällen
OEM ist die Abkürzung für Original Equipment Manufacturer (Originalausrüstungshersteller), während OBL für Own Brand Labelling (Eigenmarkenbezeichnung) steht.
Der rechtmäßige Hersteller in OEM/OBL-Fällen, in denen die EU-Rechtsordnung als Grundlage für den MDMA-Antrag ausgewählt wurde, betrachtet SFDA den Herstellernamen neben dem „Herstellersymbol“ auf dem Etikett als rechtmäßigen Hersteller. Ist dies nicht der Fall, sind der SFDA folgende Dokumente vorzulegen:
- Kopie der Konformitätsbewertungszertifikate basierend auf dem verwendeten Konformitätsbewertungsverfahren (z. B. CE-Zertifikat, DE-Zertifikat, Produktionsqualitätssicherungszertifikat usw.)
- Kopie der Konformitätserklärung des Produkts.
- Kopie der Kennzeichnung (inkl. Gebrauchsanweisung)
- Aktuellster Prüfbericht (sofern zutreffend)
Stromversorgung
Wenn das Gerät an eine Wechselstromversorgung angeschlossen ist, eine Angabe der Nennfrequenz (60 Hertz) und der Spannungswerte mit ihren Toleranzen, für die das Produkt ausgelegt ist.
IVD-Kennzeichnung
Dies zeigt an, dass das Produkt für die In-vitro-Diagnostik bestimmt ist, wenn es sich um ein IVD-Medizinprodukt handelt.
Lagerbedingungen
Gegebenenfalls ein Hinweis auf etwaige besondere Lagerungs- und Handhabungsbedingungen.
Warnung oder Vorsichtsmaßnahmen
Alle Warnungen, Vorsichtsmaßnahmen, Einschränkungen oder Kontraindikationen.
Chargen- oder Losnummer
Der Chargencode/die LOT-Nummer oder die Seriennummer ermöglichen die Anwendung von Konformitätsmaßnahmen nach der Markteinführung, wenn das Produkt nachverfolgt oder zurückgerufen werden muss. Bei Zubehörteilen von IVD-Medizinprodukten kann diese Nummer jedoch durch eine Kontrollnummer ersetzt werden, und bei Software muss sie durch eine Versionsnummer ersetzt werden.
Verfallsdatum
Eine eindeutige Angabe des Datums, bis zu dem das Produkt sicher verwendet werden kann (z. B. auf sterilen oder Einmalartikeln), sofern dies relevant ist.
Menge
Gegebenenfalls wird die Nettofüllmenge in Gewicht, Volumen, numerischer Anzahl oder einer beliebigen Kombination dieser oder anderer Begriffe angegeben, die den Verpackungsinhalt genau wiedergeben.
Zur einmaligen oder mehrfachen Verwendung
Wenn das Produkt zum einmaligen Gebrauch bestimmt ist, ist ein Hinweis auf diesen Umstand erforderlich.



