
Sobre o autor

Mohammed Jobran
Regulatory consultant with +20 years of experience working for the SFDA, multinational companies, and as a consultant in PharmaKnowl.
Navigating the SFDA medical device registration in Saudi Arabia, particularly the Medical Device Marketing Authorisation (MDMA) process, is essential for manufacturers seeking entry into the rapidly expanding Saudi market.
The MDMA requirements have evolved to cope with the advancement of international standards, such as the evolution from EU Medical Device Directives (MDD) to Medical Device Regulations (MDR). The continuous development of medical device regulations in Saudi Arabia has increased the complexity of this process.
For example, even low-risk medical devices (supplies and consumables) became in need of an authorised representative (AR) and an MDMA approval.
We at PharmaKnowl offer expert support for the registration process. Whether you’re dealing with new devices or modifying existing products, our team provides the strategic insight and technical expertise needed to achieve timely and successful market entry in Saudi Arabia.
This article clarifies the SFDA medical device registration requirements and defines the classification differences.
Para outros tipos de registro de produtos, consulte a postagem de registro SFDA .
Table of contents
Registration Application Types
A SFDA regulamenta produtos de dispositivos médicos na Arábia Saudita para garantir sua segurança, eficácia e qualidade. Os fabricantes devem obter a aprovação MDMA antes de comercializar seus produtos. Após a aprovação, eles são responsáveis por relatórios de segurança e outras operações de gerenciamento do ciclo de vida, como atualização, renovação e envio da Identificação Única do Dispositivo (UDI) .
Aqui está um resumo dos procedimentos de registro de dispositivos médicos:
MDNR (baixo risco) – cancelado
Companies used to register Non-sterile, non-measuring, Low-Risk Medical Devices through the Medical Device National Registry (MDNR), also known as “Medical Devices Listing,” which is exempt from MDMA and AR. However, in September 2022, the SFDA cancelled the MDNR procedure. Therefore, all devices must have a technical file for MDMA with an appointed AR in Saudi Arabia.
MDMA 1 (GHTF) – Cancelado
Nesta rota de aplicação, a SFDA considerou as aprovações dos países membros do GHTF, como União Europeia, Estados Unidos, Canadá, Austrália e Japão. Foi um caminho fácil de seguir, sem profundas investigações técnicas. No entanto, no final de 2021, a SFDA cancelou esta rota do GHTF.
If your product was registered initially through MDMA 1, you can still use the same route to file renewals, minor updates, or variations. But the SFDA will still request the full technical file during the process.
MDMA 2 (TFA)
From January 2022, all medical devices and in vitro diagnostics (IVDs) must receive an SFDA authorisation through the Technical File Application (TFA) registration route (MDMA2). This route resulted from the authority’s continuous regulation updates influenced by the EU MDR and IVDR. It requires presenting the technical documentation in a clear, searchable, and organised manner.
Portanto, os requisitos de registro tornaram-se mais rigorosos. Diversas condições devem ser atendidas e estudos devem ser fornecidos, como relatórios de avaliação clínica (CER), relatórios de testes de biocompatibilidade e a necessidade de conduzir um estudo de acompanhamento clínico pós-comercialização (PMCF) na Arábia Saudita.
Embora os requisitos do TFA sejam semelhantes aos da UE (marcação CE), o SFDA não exige um certificado CE. No entanto, se o arquivo do produto não possuir a marca CE, ele será submetido a uma avaliação rigorosa.
Requisitos de registro
A seguir, um resumo dos requisitos, que variam de acordo com o tipo e a classe do produto. Aqui estão as seções gerais do arquivo de registro:
- Administrative & Product Identification
- Table of contents
- Descrição do dispositivo
- Uso/finalidade pretendida
- Device Classification
- Model, Sizes, Accessories, and Variants
- Device label and Packaging Information
- Instructions for use (IFU)
- Informações de design
- Informações de fabricação
- Device History (Approvals from Reference Countries)
- Safety, Risk & Performance Documentation
- Essential Principles (EP List) of Safety & Performance (Required checklist)
- Análise de benefício-risco
- Arquivo de Gestão de Riscos (Plano e Relatório)
- Preclinical testing data
- Biocompatibility Test Reports.
- Electrical Safety, EMC, Software validation (as applicable)
- Sterilisation and Shelf-Life Validation (if sterile)
- Clinical Evaluation & Surveillance
- Plano e relatório de investigação clínica
- Relatório de Avaliação Clínica (CER)
- Acompanhamento Clínico Pós-Comercialização (PMCF)
- Vigilância Pós-Mercado (PMS), Plano e Relatório
- Relatório Periódico de Atualização de Segurança (PSUR) para dispositivos médicos.
Download: Checklist of SFDA Medical Device Required Files for MDMA application
Processo de Registro
Time needed: 90 days
Podemos resumir o processo de aprovação de registro de dispositivo médico (MDMA) nas seguintes etapas:
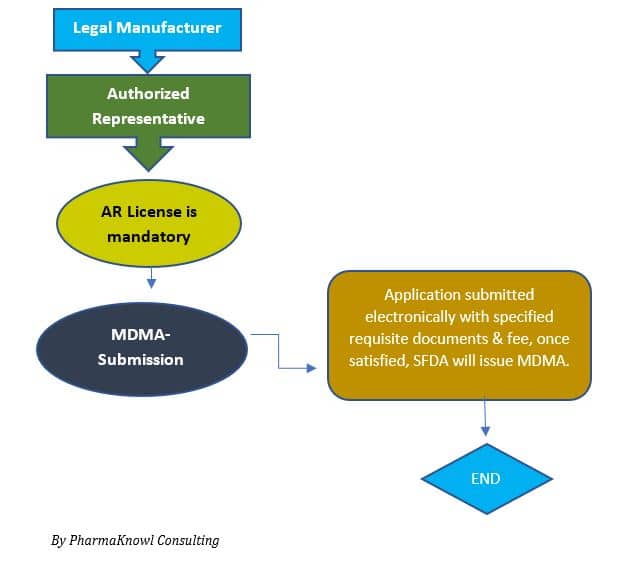
- Nomeação de um Representante Autorizado (AR)
Manufacturers must appoint an AR to represent their company in Saudi Arabia. An AR license will be issued by SFDA, after which the AR can apply for the device registration.
- Análise de Lacunas
Applicant must establish a thorough gap analysis of the registration file vs. all applicable requirements. Identifying the gaps is essential to determining the relevant studies and tests, such as Stability, CER, Biocompatibility, risk management, and PMCF.
- Submissão ao MDMA
O requerente envia o arquivo técnico ao SFDA .
- Validação
SFDA garante boas práticas de submissão sem avaliar seu conteúdo.
- Pagamento de taxas
The applicant will receive an SFDA invoice to pay before the start of the assessment.
- Avaliação de Aplicação
The SFDA evaluators will review the file and send back their inquiries in multiple waves.
- Aprovação de MDMA
The legal manufacturers receive an MDMA certificate that permits them to market the product in Saudi Arabia.
Classificação
Applicants must classify their products according to the SFDA medical device classification rules within the MDMA application. These rules correspond to the EU regulation (CE certificate) since they are the unofficial SFDA reference regulations, but do not necessarily match them.
Dispositivo médico
SFDA medical device classification is either Class A, B, C, or D, based on the risk class. The class is necessary to determine the registration procedure and its requirements, as discussed in the table below:
| Classificação SFDA | Classe de Risco | Regra de Classificação MDR |
| Classe A | Baixo | EU |
| Classe A – Estéril | Baixo-médio | É |
| Classe A – Função de medição | Baixo-médio | Eu sou |
| Classe A – Instrumentos cirúrgicos reutilizáveis | Baixo-médio | Ir |
| Classe B | Baixo-médio | IIa |
| Classe C | Médio-alto | IIb |
| Classe D | Alto | III |
A classificação é feita da seguinte forma:
- Uso pretendido.
- Nível de risco (probabilidade e gravidade do dano aos pacientes, usuários e outros).
- Nature of the device on or within the human body (e.g. invasive)
- Use duration (transient, short-term, long-term)
Identical devices could be classified differently according to the targeted body part or clinical setting. Therefore, the intended use is crucial in determining the correct classification. The intended use reference is reflected in the following:
- Instruções de uso (IFU)
- Rótulo
- Materiais promocionais
- Ficha Técnica
In Vitro Diagnostics (IVDs)
For in vitro diagnostic medical devices, the (SFDA) uses a risk-based classification system broadly aligned with the International Medical Device Regulators Forum (IMDRF) principles and is similar in structure to the EU IVDR, though applied independently.
| Classificação SFDA IVD | Classe de Risco | Regra de Classificação |
| Classe A | Baixo risco individual e baixo risco para a saúde pública | UM |
| Classe B | Risco individual moderado e/ou baixo risco para a saúde pública | B |
| Classe C | Alto risco individual e/ou risco moderado para a saúde pública | C |
| Classe D | Alto risco individual e alto risco para a saúde pública | D |
O requerente é responsável por determinar a classe do IVD por:
- Aplicação das regras de classificação para dispositivos médicos de diagnóstico in vitro (DIV), e
- Considerando:
- Uso pretendido
- Nível de risco
Bundling Multiple Devices
Grouping multiple devices in the same application is permissible; the applicant may bundle up to 50 items in one MDMA application. However, products must meet several conditions, such as having the same legal manufacturer, the same risk class, and the same intended use.
| Bundling Type | Criteria | Examples |
| Single Product | A device sold in multiple sizes or models with the same intended use | Syringes with different volumes |
| Family | Devices with the same risk class, manufacturer, design, and intended use | Catheters of varying lengths |
| System | Devices intended to operate together for a common purpose | ECG machine + electrodes + leads |
| Set | Devices packaged together and intended for a common medical purpose | Surgical kits or dental kits |
| Software Suite | Related software modules that work as part of the same platform | Hospital management or imaging analysis software |
| IVD’s | Can be bundled in case of the same risk class, manufacturer, design, and intended use | Blood collection tubes, Culture media |
Cronogramas
The assessment timelines vary according to many factors, such as risk class, complexity of the product, number of bundled products, and the completeness of the file. Unidentified gaps before the submission may delay the approval. The entire timeline for the registration project includes:
- Gap Analysis Time
This part depends on the number and type of gaps, the speed of manufacturer feedback, and the regulatory staff’s experience. - Tempo de avaliação SFDA
Para os cronogramas oficiais de revisão, consulte nosso artigo Cronogramas SFDA .
Tarifas
As taxas de solicitação de MDMA variam de acordo com a classe de risco do dispositivo médico e o número de produtos na solicitação. Para mais informações, consulte nossa publicação atualizada sobre Taxas SFDA .
Renovação
A validade padrão do MDMA é de três anos. Quando a licença estiver próxima do vencimento, os fabricantes podem enviar um pedido de renovação do MDMA até três meses (90 dias) antes da data de vencimento. Se o registro for feito no aplicativo obsoleto MDMA1 (GHTF), o processo de renovação será longo e exigirá requisitos complexos, assim como um novo registro. Em contrapartida, a renovação do MDMA2 será mais rápida.
Atualizar
Para fazer alterações no dispositivo médico registrado, os fabricantes devem enviar uma solicitação de atualização de MDMA .
Certificado MDMA
SFDA emitirá um certificado MDMA em árabe e inglês contendo o seguinte:
- Informações do fabricante (titular da licença)
- Nome do dispositivo médico e informações do grupo de dispositivos médicos.
- Período de validade
- Número do certificado

Categorias de produtos
Aqui estão alguns exemplos de tipos e categorias de dispositivos médicos na Arábia Saudita:
- Dispositivos de diagnóstico:
- Equipamentos de imagem: aparelhos de raio X, tomógrafos computadorizados.
- Dispositivos de monitoramento: aparelhos de ECG e monitores de pressão arterial.
- Dispositivos de diagnóstico in vitro: testes de gravidez, medidores de glicemia.
- Dispositivos terapêuticos:
- Aparelhos respiratórios: Ventiladores, nebulizadores.
- Dispositivos de infusão: bombas de insulina e equipamentos de terapia intravenosa.
- Dispositivos de reabilitação: cadeiras de rodas, próteses.
- Dispositivos implantáveis:
- Dispositivos cardiovasculares: marcapassos, stents.
- Implantes ortopédicos: substituições de quadril, hastes para coluna, substituições de articulações.
- Instrumentos cirúrgicos:
- Instrumentos de corte: bisturis, tesouras cirúrgicas, fórceps.
- Grampeadores e Suturas: Grampeadores cirúrgicos, suturas absorvíveis.
- Dispositivos Dentários:
- Materiais restauradores: coroas dentárias, obturações.
- Aparelhos ortodônticos: aparelhos, contenções.
- Outros: brocas odontológicas
- Dispositivos Oftálmicos:
- Lentes corretivas: Lentes de contato, óculos.
- Equipamentos cirúrgicos: máquinas LASIK, lentes intraoculares.
- Software Médico (SaMD) :
- Software de diagnóstico: IA para análise de imagens.
- Software de planejamento de tratamento: ferramentas de planejamento de radioterapia.
- Equipamentos de laboratório:
- Analysers: Blood gas analysers and haematology analysers.
- Sterilisation equipment: Autoclaves, UV sterilisers.
- Gases Medicinais
- Dispositivos de assistência médica domiciliar:
- Dispositivos de monitoramento: monitores de glicemia e termômetros digitais.
- Auxílios de mobilidade: andadores, patinetes elétricos.
- Dispositivos terapêuticos (por exemplo, máquinas de diálise)
Licença de Importação de Dispositivos Médicos (MDIL)
Importing medical devices requires a valid MDMA. However, the SFDA may exempt some medical devices from registration and only require them to issue a medical device importing license (MDIL). For example:
- Demonstration or training purposes, medical devices.
- Produtos químicos (produtos acabados), sejam eles classificados como dispositivos médicos ou utilizados em conjunto com dispositivos médicos (por exemplo, gases utilizados para calibrar dispositivos médicos, bem como produtos químicos utilizados para garantir a operação de esterilização de dispositivos médicos, a fabricação de próteses e a preservação de tecidos ou células). Excluem-se os seguintes produtos (se o produto químico for classificado como dispositivo médico e o dispositivo tiver que conter MDMA).
- Materiais radioativos
- IVD médico
- IVD não médico
- Precursores químicos.
- Aparelhos de destilação para profissionais de saúde ou instituições educacionais.
- Produtos para uso educacional e de pesquisa.
- Dispositivos/suprimentos médicos semiacabados (e produtos químicos brutos e não brutos) para fabricação local (a fabricação inclui reforma, montagem, embalagem e etiquetagem).
Registro de empresa
The SFDA also regulates medical device companies, enforcing different requirements for local and international companies to receive an SFDA license to manufacture, import, or distribute medical devices. Below are the requirements for both types:
Empresa saudita local
The SFDA licenses local medical device companies such as importers, distributors, warehouses, authorized representatives, and manufacturers. The local company license name is the Medical Device Establishment License (MDEL). All previous types of companies must implement a quality management system (QMS) and have an ISO 13485 certificate.
Fabricante Legal
International legal manufacturers must appoint an authorised representative (AR) in Saudi Arabia; this is the first step in enabling communication with the SFDA. The AR’s primary responsibility is product compliance in pre- and post-marketing activities. The AR will maintain registration status, facilitate shipments, monitor safety, report cases, and represent the company in regulatory or legal matters.
Serviço de Registro
Whether your company is new to or already active in the Saudi market, PharmaKnowl will help you comply with the SFDA and facilitate your business by providing compliance, insights, and supply management to your distributors without shipment commission.
PharmaKnowl is an SFDA-licensed service provider in Saudi Arabia representing well-known international companies. Contact us to schedule an exploratory meeting or request more information.
Frequently Asked Questions
MDMA is the official approval issued by the Saudi Food and Drug Authority (SFDA) that allows a medical device or IVD to be marketed in the Kingdom. It confirms that the product meets all local regulatory, safety, and performance requirements based on a technical file review.
The MDMA certificate is valid for three years. Renewal can be initiated up to 90 days before expiration.
Yes. For international manufacturers, appointing an SFDA-licensed Authorised Representative (AR) is mandatory to submit the MDMA application and manage post-market obligations in Saudi Arabia.
The typical MDMA timeline is 35 business days if all requirements are fulfilled, but the timeline can vary due to technical file gaps, product complexity, or missing documentation.
Yes. A maximum number of 50 medical devices can be grouped/bundled into a single MDMA application as long as they meet bundling criteria.
The company is not allowed to market its product when the certificate expires. The company must renew as soon as possible and ensure its AR license is valid.
While prior approval (e.g., CE mark, FDA 510(k)) can support your application, the SFDA conducts an independent evaluation. MDMA approval is not granted automatically based on foreign registrations. SFDA does not require CE or FDA approval as long as the full technical file is available according to SFDA requirements.
MDMA applies to the product registration certificate for a medical device or IVD in KSA, while MDEL is the importer or distributor license.
MDMA-1: Registration route based on approvals from Global Harmonisation Task Force (GHTF) countries: EU, US, Canada, Australia, Japan. It was easier and faster without the need for complete technical files. However, this route was cancelled by SFDA and replaced with MDMA 2.
MDMA 2, which is the current registration route (since 2022). It requires complete Saudi-specific technical files (similar to EU MDR standards) and undergoes a more stringent SFDA evaluation. It does not require any prior GHTF approvals.



