Medical device products in Saudi Arabia must obtain approval from the Saudi Food and Drug Authority (SFDA) before marketing. Manufacturers and their authorized representatives must submit a request for approval through the Medical Device Marketing Authorization (MDMA) application.
In this post, we explain how to obtain MDMA approval and outline the relevant laws and regulations for medical devices.
Table of contents
Introduction
The SFDA is a member of the International Medical Device Regulators Forum (IMDRF), and Saudi Arabia’s medical device regulations are similar to those in the EU under the MDR. The Saudi authority regulates related establishments, such as manufacturers and warehouses in addition to product regulations, to ensure their safety, efficacy, and quality. Products that pass the review are granted an MDMA certificate.
On the other hand, once products are approved, the authority enforces post-marketing obligations on the legal manufacturers, including safety reporting, Unique Device Identification (UDI), and other life-cycle management processes.
Application Types
There were previously several types of SFDA medical device registration applications. However, most are now sunsetted. The authority unified all into a single route: the MDMA application.
MDNR (Low Risk) – Canceled
Companies used to register Non-sterile, non-measuring, Low-Risk Medical Devices through the Medical Device National Registry (MDNR), also known as “Medical Devices Listing,” which is exempt from MDMA and AR. However, in September 2022, the SFDA canceled the MDNR procedure. Therefore, all devices must have a technical file for MDMA with an appointed AR in Saudi Arabia.
MDMA 1 (GHTF) – Canceled
In this application route, the SFDA considered approvals from GHTF member countries, including the European Union, the United States, Canada, Australia, and Japan. It was an easy route to follow, with no deep technical inquiries required. However, by the end of 2021, the SFDA had canceled this GHTF route.
MDMA 2 (TFA) – Active
From January 2022, MDMA2 became the only option for all medical devices and in vitro diagnostics (IVDs). It requires submission of a complete Technical File Application (TFA). Note that the CE certificate is not required.
Registration Requirements
The requirements vary by product type and class. Here are a few examples:
- Administrative & Product Identification
- Device Classification
- Device Label and Packaging Information
- Safety, Risk & Performance Documentation
- Essential Principles (EP List) of Safety & Performance
- Benefit-risk analysis
- Risk Management File (Plan & Report)
- Preclinical testing data
- Biocompatibility Test Reports
- Electrical Safety, EMC, Software validation (as applicable)
- Sterilization and Shelf-Life Validation (if sterile)
- Clinical Evaluation & Surveillance
- Clinical investigation plan, and Clinical Evaluation Report (CER)
- Post-Market Clinical Follow-Up (PMCF), Post-market Surveillance (PMS), and Periodic Safety Update Report (PSUR).
Download the complete list: MDMA Checklist
Registration Process
Time needed: 90 days
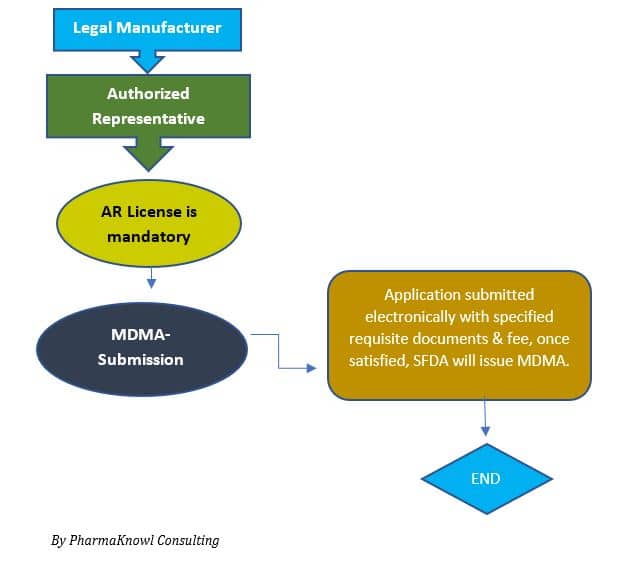
We can summarise the MDMA approval process in the following few steps:
- Appointing an Authorized Representative (AR)
Manufacturers appoint an AR to represent their company in Saudi Arabia.
- Gap Analysis
The applicant must conduct a thorough gap analysis of requirements, relevant studies, and tests, including Stability, CER, Biocompatibility, risk management, and PMCF.
- MDMA Submission
The applicant submits the technical file to the SFDA.
- Validation
SFDA ensures good submission practice without assessing its content.
- Fees Payment
The applicant will receive an SFDA invoice to pay before the assessment begins.
- Application Assessment
SFDA reviews the file and sends inquiries.
- MDMA Approval
The legal manufacturers receive an MDMA certificate.
Classification
Applicants must classify their products in accordance with the SFDA rules within the MDMA application. These rules correspond to the EU regulation (CE certificate). In the sections below, we list the risk classifications for devices and IVDs in Saudi Arabia.
Medical Device Classes
SFDA medical device classification is either Class A, B, C, or D, based on the risk class. The class is necessary to determine the registration procedure and its requirements, as discussed in the table below:
| SFDA Classification | Risk Class | MDR Classification Rule |
| Class A | Low | I |
| Class A | Low-medium | Is |
| Class A | Low-medium | Im |
| Class A | Low-medium | Ir |
| Class B | Low-medium | IIa |
| Class C | Medium-high | IIb |
| Class D | High | III |
Classification is according to the following:
- Intended use.
- Risk level (probability and severity of harm to patients, users, and others).
- Nature of the device on or within the human body (e.g., invasive / non-invasive)
- Use duration (transient, short-term, long-term)
Identical devices could be classified differently according to the targeted body part or clinical setting. Therefore, the intended use is crucial in determining the correct classification. The intended use reference is reflected in the following:
- Instructions for Use (IFU)
- Promotional materials
- Technical File
In Vitro Diagnostics (IVDs) Classes
For in vitro diagnostic medical devices, the SFDA employs a risk-based classification system that is broadly aligned with the International Medical Device Regulators Forum (IMDRF) principles and is similar in structure to the EU IVDR, although it is applied independently.
| SFDA IVD Classification | Risk Class | IVDR Classification Rule |
| Class A | Low individual risk and low public health risk | A |
| Class B | Moderate individual risk and/or low public health risk | B |
| Class C | High individual risk and/or moderate public health risk | C |
| Class D | High individual risk and high public health risk | D |
The applicant is responsible for determining the class of the IVD by:
- Applying the classification rules for IVD medical devices, and
- Considering:
- Intended use
- Level of risk
Bundling Multiple Devices
Grouping multiple devices in the same application is permissible; the applicant may bundle up to 50 items in one MDMA application. However, products must meet several conditions, such as having the same legal manufacturer, risk class, and intended use.
| Bundling Type | Criteria | Examples |
| Single Product | A device sold in multiple sizes or models with the same intended use | Syringes with different volumes |
| Family | Can be bundled if the devices have the same risk class, manufacturer, design, and intended use | Catheters of varying lengths |
| System | Can be bundled if the devices are intended to operate together for a common purpose | ECG machine + electrodes + leads |
| Procedure Pack / Kits | Can be bundled if the devices are packaged together and intended for a common medical purpose | Surgical kits or dental kits |
| Software Suite | Related software modules that work as part of the same platform | Hospital management or imaging analysis software |
| IVD’s | Can be bundled in case of the same risk class, manufacturer, design, and intended use | Blood collection tubes, Culture media |
Timelines
The assessment timelines vary according to several factors, including risk class, product complexity, the number of bundled products, and the completeness of the file. Unidentified gaps before the submission may delay the approval. The entire timeline for the registration project includes:
- Gap Analysis Time
This part depends on the number and type of gaps, the speed of manufacturer feedback, and the regulatory staff’s experience. - SFDA Assessment Time
For the official review timelines, refer to our article SFDA timelines.
Fees
The MDMA application fees vary by medical device risk Class and the number of products in the application. For more information, refer to our updated SFDA Fees post.
Renewal
MDMA’s default validity is three years. Manufacturers can submit an MDMA renewal application up to six months (180 days) before the expiration date.
Update
To make changes to the registered medical device, manufacturers must submit an MDMA update application.
MDMA Certificate
SFDA will issue an MDMA certificate in both Arabic and English containing the following:
- The manufacturer’s information (License Holder)
- Medical Device Name
- Validity Period
- Certificate number
Note: The name of the Authorized Representative or the local importer/distributor is not mentioned nor linked to the license.

Product Categories
Here are examples of medical device types and categories in Saudi Arabia:
- Diagnostic Devices:
- Imaging Equipment: X-ray machines, CT scanners.
- Monitoring Devices: ECG machines and blood pressure monitors.
- In Vitro Diagnostic Devices: Pregnancy tests, blood glucose meters.
- Therapeutic Devices:
- Respiratory Devices: Ventilators, nebulizers.
- Infusion Devices: Insulin pumps and IV therapy equipment.
- Rehabilitation Devices: Wheelchairs, prosthetics.
- Implantable Devices:
- Cardiovascular Devices: Pacemakers, stents.
- Orthopedic Implants: Hip replacements, spinal rods, joint replacements.
- Surgical Instruments:
- Cutting / Grasping Instruments: Scalpels, surgical scissors, forceps.
- Staplers and Sutures: Surgical staplers, absorbable sutures.
- Dental Devices:
- Restorative Materials: Dental crowns, fillings.
- Orthodontic Appliances: Braces, retainers.
- Other: dental drills
- Ophthalmic Devices:
- Corrective Lenses: Contact lenses, eyeglasses.
- Surgical Equipment: LASIK (Laser Surgical) machines, intraocular lenses.
- Medical Software (SaMD):
- Diagnostic Software: AI for image analysis.
- Treatment Planning Software: Radiation therapy planning tools.
- Laboratory Equipment:
- Analyzers: Blood gas analyzers and hematology analyzers.
- Sterilization equipment: Autoclaves, UV sterilizers.
- Medical Gases
- Home Healthcare Devices:
- Monitoring Devices: Blood glucose monitors and digital thermometers.
- Mobility Aids: Walkers, electric scooters.
- Therapeutic devices (e.g., dialysis machines)
Medical Device Importing License (MDIL)
Importing medical devices requires a valid MDMA. However, the SFDA may exempt some medical devices from registration and require only that they obtain a medical device importing license (MDIL). For example:
- Demonstration or training purposes, medical devices.
- Chemicals (finished products), whether classified as medical devices or used in conjunction with medical devices (e.g., gases used to calibrate medical devices, as well as chemicals used to ensure the sterilization operation of medical devices, the manufacturing of prostheses, and the preservation of tissues or cells). Excluding the following products:
- Radioactive materials
- Medical IVD
- Non-medical IVD
- Chemical precursors.
- Distillation apparatuses for healthcare providers or educational facilities.
- Research and Educational use products.
- Semi-finished medical devices/supplies (and raw and non-raw chemicals) for local manufacturing (manufacturing includes refurbishing, assembling, packaging, and labeling).
Establishment Registration
The SFDA regulates medical device companies, including manufacturers, authorized representatives, importers, and distributors.
Local Saudi Company
The SFDA licenses local medical device importers, distributors, warehouses, authorized representatives, and manufacturers. They must hold an approved Medical Device Establishment License (MDEL) and operate under an ISO 13485-certified quality management system.
International Manufacturers
International legal manufacturers looking to export their products to Saudi Arabia must appoint an authorized representative (AR).
Registration Service
Whether your company is new to the Saudi market or already established, PharmaKnowl will help you comply with the SFDA and facilitate your business by providing compliance, insights, and supply management to your distributors, all without requiring shipment commissions.
PharmaKnowl is an SFDA-licensed service provider in Saudi Arabia representing well-known international companies. Contact us to schedule an exploratory meeting or request more information.
Frequently Asked Questions
MDMA is the official approval issued by the Saudi Food and Drug Authority (SFDA) that allows a medical device or IVD to be marketed in the Kingdom. It confirms that the product meets all local regulatory, safety, and performance requirements based on a review of the technical file.
The MDMA certificate is valid for three years.
Yes. For international manufacturers, appointing an SFDA-licensed Authorized Representative (AR) is a must before they can submit the MDMA and manage post-market obligations in Saudi Arabia.
The typical MDMA timeline is 35 business days if all requirements are met, but it can vary due to technical file gaps, product complexity, or missing documentation.
Yes. A maximum number of 50 medical devices can be grouped/bundled into a single MDMA application as long as they meet bundling criteria.
The company is not allowed to market its product when the certificate expires. The company must renew its AR license as soon as possible to ensure it remains valid.
While prior approval (e.g., CE mark, FDA 510(k)) can support your application, the SFDA conducts an independent evaluation. MDMA approval is not granted automatically based on foreign registrations. SFDA does not require CE or FDA approval, provided the complete technical file is available in accordance with SFDA requirements.
MDMA applies to the product registration certificate for a medical device or IVD in KSA, while MDEL is the importer or distributor license.
MDMA-1: Registration route based on approvals from Global Harmonization Task Force (GHTF) countries: EU, US, Canada, Australia, Japan. It was easier and faster without the need for complete technical files. However, this route was canceled by SFDA and replaced with MDMA 2.
MDMA 2, which is the current registration route (since 2022). It requires complete Saudi-specific technical files (similar to EU MDR standards) and undergoes a more stringent SFDA evaluation. It does not require any prior GHTF approvals.
About the Author

Regulatory affairs consultant, with more than 20 years of experience working for the SFDA and the industry.
Resources




Services
Events