SFDA requires all medical device companies in Saudi Arabia to comply with the Unique Device Identification (UDI) regulations. They must register their product’s UDI information in the SFDA UDI system. The rules are gradually enforced based on the device risk class and can only be submitted through the authorised representatives in Saudi Arabia after the medical device approval.
Table of contents
What is UDI?
Unique Device Identification UDI is a series of numeric or alphanumeric characters generated through a globally accepted coding system. It is an identifier that unambiguously identifies a device through its distribution and use. Comprised of Two Parts:
1. A Device Identifier (DI): a mandatory, fixed portion of a UDI that identifies the specific Product Number of a device and the labeller of that device.
2. A Production Identifier (PI): a conditional, variable portion of a UDI that identifies one or more of the following when included on the label of the device:
- The lot or batch within which a device was manufactured.
- The serial number of a specific device.
- The expiration date of a specific device.
- The manufacturing date of a specific device.
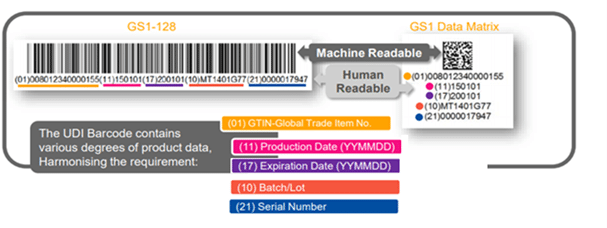
The UDI shall be placed on the label of the device and the higher levels of the packaging materials, and be presented in two forms:
- Easily readable plain-text (also known as HRI-Human Readable Interpretation), and
- AIDC technology (Automatic Identification and Data Capture).
The UDI Barcode- What is on the Label?
Scope
- All medical devices and their accessories, except those custom-made for specific patients, and investigational as well as research-use devices
- Manufacturers, authorised representatives
Recognised Issuing Agencies
UDI-recognised issuing Agencies are the following entities: GS1, HIBCC, and ICCBBA.
UDI Database
- The manufacturer or its authorised representative shall submit and maintain the appropriate data in the UDI database.
- Shall be checked and maintained periodically by the manufacturer
- The data for the new UDI-DI shall be available in the database when the device is placed on the market.
- All specified (non-private) data in the database will be publicly available.
- All data in GHAD and Saudi-DI, such as model name and GMDN code, are accurate and valid.
WHY UDI?
SFDA’s UDI System aims to standardise medical device identification (and their accessories). The UDI system seeks to increase patient safety and optimise patient care by facilitating the following:
Increasing patient safety
- Increase patient safety: In adverse event reports and other post-market surveillance activities, field safety corrective actions.
- Improving identification and traceability: Control at ports and throughout its life cycle, including identification and documentation at the point of patient use.
- Identification of counterfeits.
- Safe and effective use of devices and reduction of medical errors.
- Patient’s electronic health records.
Utilised in other aspects
- Purchasing and inventory management
- Medical insurance activities
- Cost control & monitoring
- Replace in-house coding with UDI
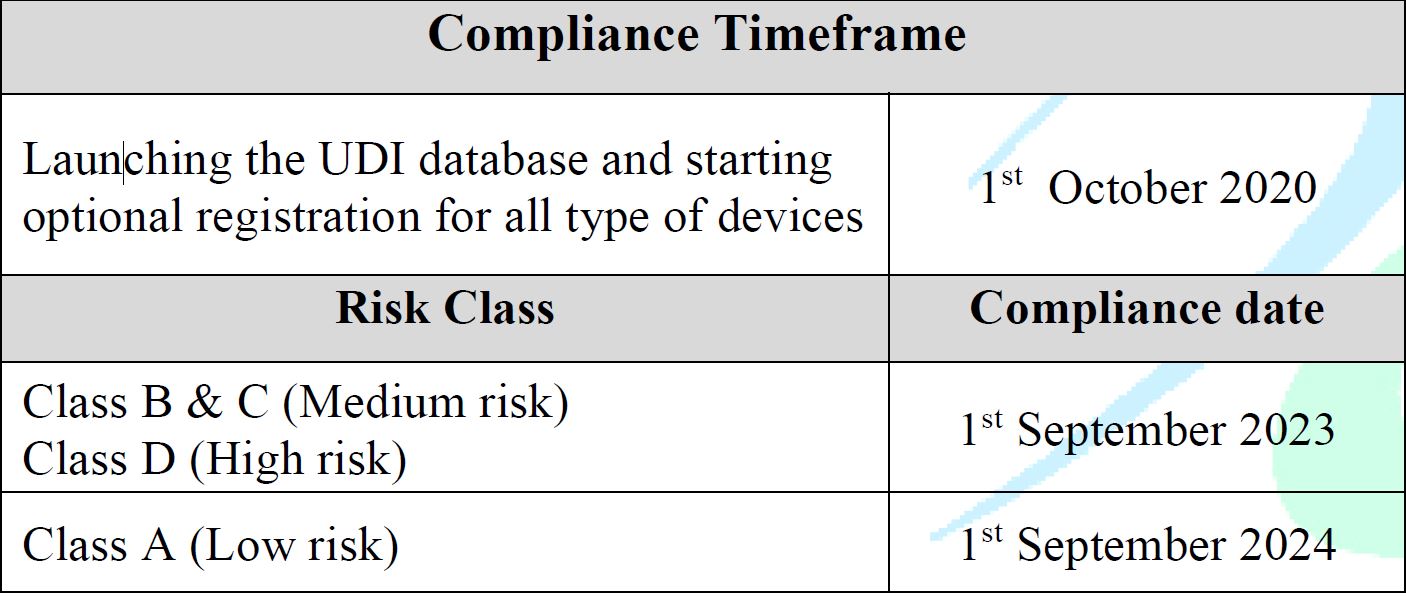
Effective Dates
- Effective from 1st Oct 2020
Launching the UDI database and starting optional registration for all types of devices. - The enforcement plan will be based on the product’s risk class as follows:
- High Risk (D): 1 September 2023
- Medium Risk (B, C): 1 September 2023
- Low Risk (Class A): 1 September 2024
Who can register UDI?
Only Saudi-authorised representatives can access the SFDA UDI platform. Contact us for support.
Read More:
About the Author

Published by regulatory affairs team in PharmaKnowl, Riyadh office.
Resources




Services
Events